Computational Biology
(BIOSC 1540)
Oct 31, 2024
Lecture 17:
Docking and virtual screening
Announcements
-
A06 is due tonight by 11:59 pm
- Reminder: There is a (soft) limit of 100 words for each question
- A07 (final assignment) will be released tomorrow
- No class on Tuesday (Nov 5) for election day
- The next exam is on Nov 14
- We will have a review session on Nov 12
- Request DRS accommodations if needed
- Project will be released Nov 21 and is due Dec 10

After today, you should have a better understanding of
Need limitations of alchemical simulations
Alchemical simulations: Precise but expensive
Alchemical simulations compute free energy changes by gradually transforming one molecule into another
Importance in drug discovery: Highly precise, offering detailed insights into binding affinities essential for drug design

Why are alchemical simulations computationally expensive?
Atomistic forces: Computes forces for all atoms in proteins, ligands, cofactors, ions, solvents for millions of structures
Detailed sampling: Captures a wide range of conformations, which adds more dimensions to the calculation
Alchemical parameters: Simulations must be performed at various alchemical parameters
Okay, so how long is this really?
~10,000 CPU hours
- ~417 days on a 1 core
- ~42 days on 10 cores
- ~4 days on 100 cores
- ~10 hours on 1,000 cores
(For context, most supercomputers have ~24 cores per $30,000 node.)
Efficient methods are needed for virtual screening
Remember: Chemical space is unfathomably large and the role of computation is to virtually test as many compounds as possible
Value of data-driven approaches for drug discovery
After today, you should have a better understanding of
Accurate and efficient binding predictions are essential
Objective: Directly predict binding affinity from protein and ligand structures with high accuracy and minimal computational resources.
We can carefully simplify our methodology to improve speed with (hopefully) minimal impact to accuracy
What are some ideas?

Avoid sampling all microstates and determine one "optimal" protein-ligand structure
Using this bound structure, predict a "score" that is correlated to binding affinity
This is called docking
Docking simplifies the binding free energy prediction problem to enhance speed
Identifying relevant protein conformations
After today, you should have a better understanding of
Accurate, reproducible docking requires a relevant protein conformation
Significance of Protein Conformation in Docking
- Protein-ligand interactions are highly dependent on the protein's 3D structure.
- Using an inappropriate protein conformation can lead to inaccurate docking results.
Docking still considers the protein structure, but we only select one
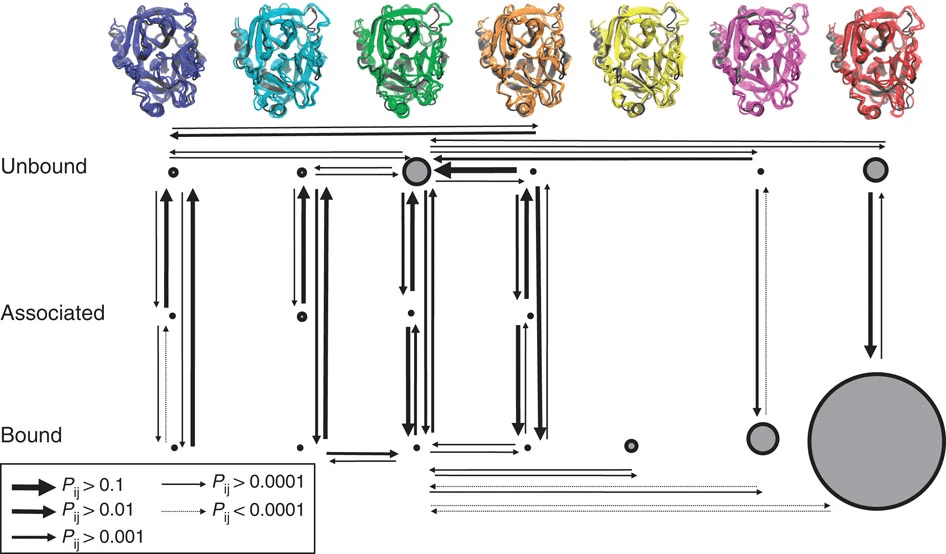
Challenge: Proteins Are Dynamic Molecules
- Conformational Flexibility: Proteins are not rigid structures; they exhibit movements ranging from side-chain rotations to large domain motions
- Impact on Binding Sites: The shape and properties of the binding site can change, affecting ligand binding affinity and specificity.
- Limited Experimental Structures: Crystallography and NMR provide snapshots of protein conformations but may not capture all relevant states.
Sources of Protein Conformational Data
Experimental Methods
- Molecular Dynamics (MD) Simulations: Explore the conformational space over time.
- Normal Mode Analysis (NMA): Identifies collective motions in proteins.
- Ensemble Generation Methods: Generate multiple protein conformations for docking.
- X-ray Crystallography: Provides high-resolution structures but may miss dynamic conformations.
- NMR Spectroscopy: Captures ensembles of conformations but is limited to smaller proteins.
Computational Techniques
Experimental Structure Selection Criteria
-
Resolution and Quality
- Prefer structures with higher resolution (e.g., <2.5 Å).
- Assess reliability using R-factors and validation reports.
-
Ligand-Bound vs. Apo Structures
-
Ligand-Bound (Holo) Structures
- Provide direct insight into binding site conformation.
-
Apo Structures
- May reveal binding site flexibility in the absence of ligands.
-
Ligand-Bound (Holo) Structures
-
Relevance to Target Ligand
- Choose structures co-crystallized with ligands similar to those of interest.
Molecular Dynamics Simulations for Conformational Sampling
Extract representative structures using clustering algorithms.

Identify conformations with open or closed binding sites.
Importance of Water Molecules
Role in Binding: Structured water molecules can mediate interactions between the protein and ligand.
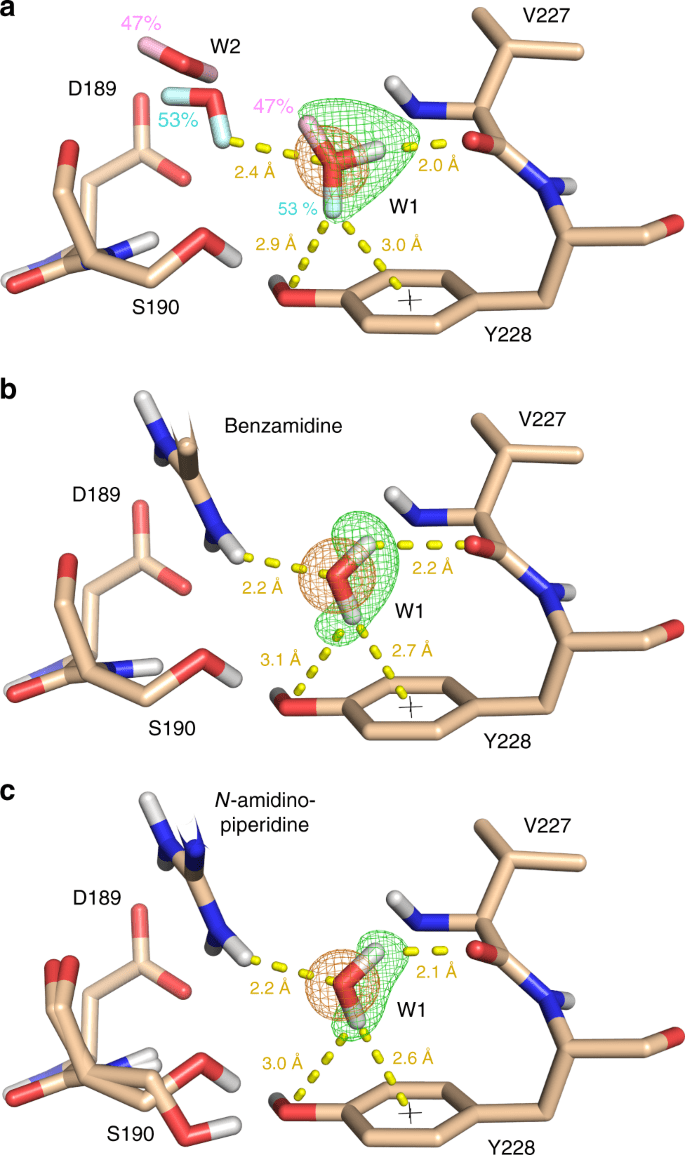
Handling Water in Docking
- Some docking programs allow explicit water molecules in the binding site.
- Alternatively, consider their effect implicitly in scoring functions.
Inclusion Criteria: Retain water molecules that are conserved across multiple crystal structures.
Detecting binding pockets
After today, you should have a better understanding of
Accurate Binding Pocket Detection is Crucial for Docking
The binding pocket is the specific region where a ligand interacts with a protein
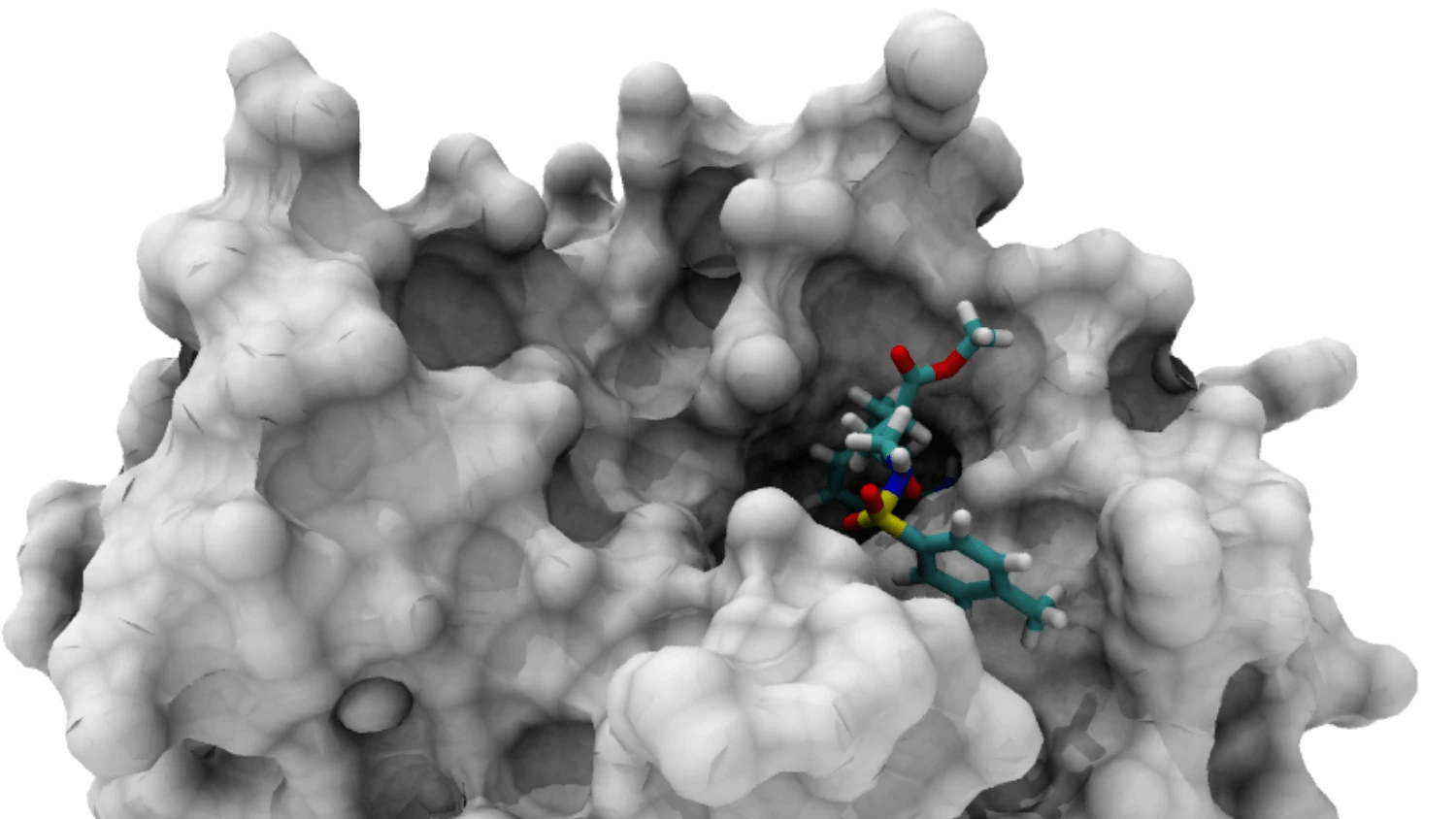
Accurate identification of binding pockets is essential for successful docking and virtual screening.
Understanding Protein Surface Topography
Terminology
- Binding Pocket: A cavity that can accommodate a ligand.
- Active Site: The functional region where biochemical reactions occur (often a binding pocket in enzymes).
Protein Surface Characteristics
- Convex Regions: Typically inaccessible to ligands.
- Concave Regions (Cavities): Potential binding sites.
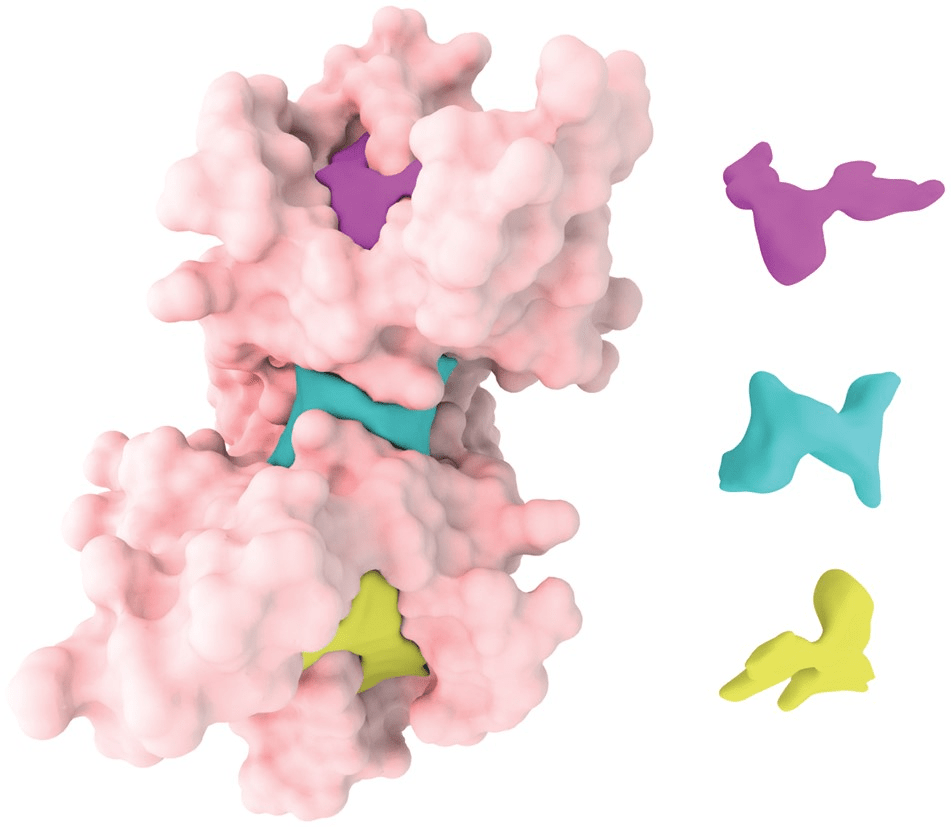
Classification of Binding Pockets
- Orthosteric Sites: The primary active site where endogenous ligands bind.
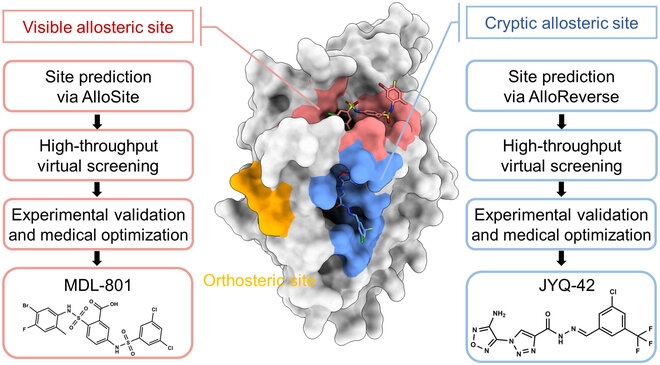
- Allosteric Sites: Secondary sites that modulate protein function upon ligand binding.
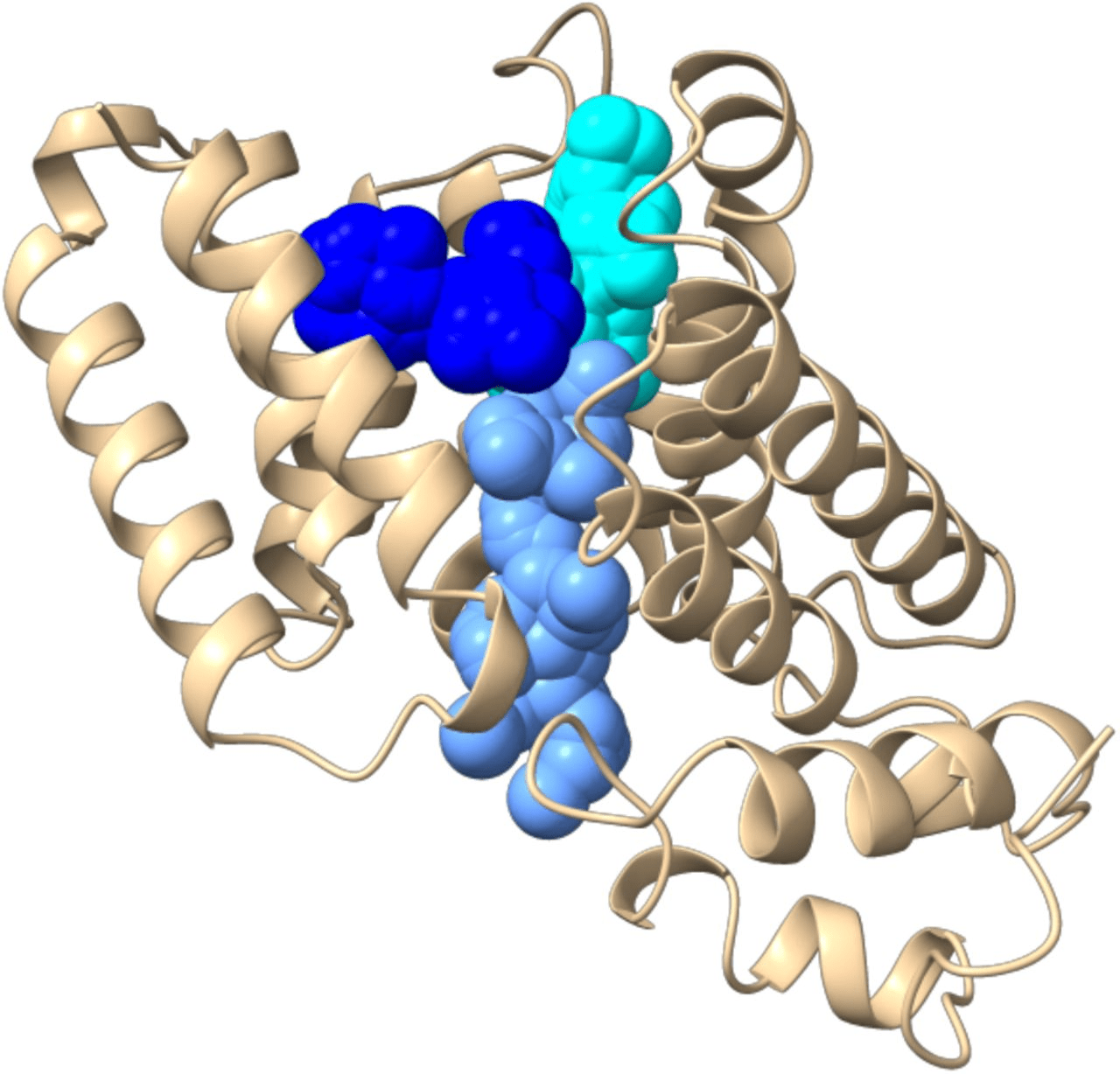
- Cryptic Sites: Binding pockets not apparent in the unbound protein structure but form upon ligand binding or conformational change.
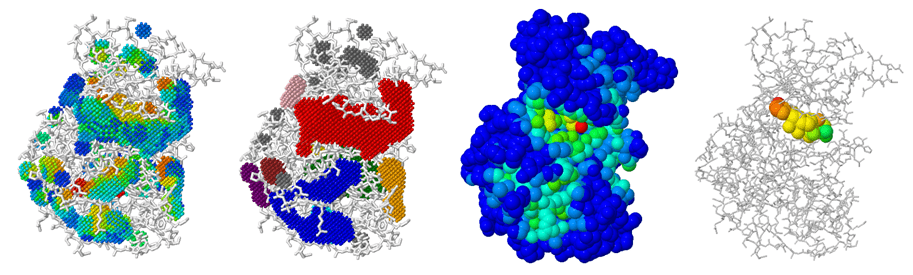
Geometry-Based Pocket Detection Techniques
Alpha Shape Theory: Uses Delaunay triangulation and alpha complexes to define cavities.

Grid-Based Pocket Detection
-
Methodology
- Overlay a 3D grid on the protein structure.
- Classify grid points as inside, outside, or on the surface.
-
Pocket Identification
- Clusters of surface grid points forming concave regions indicate potential pockets.
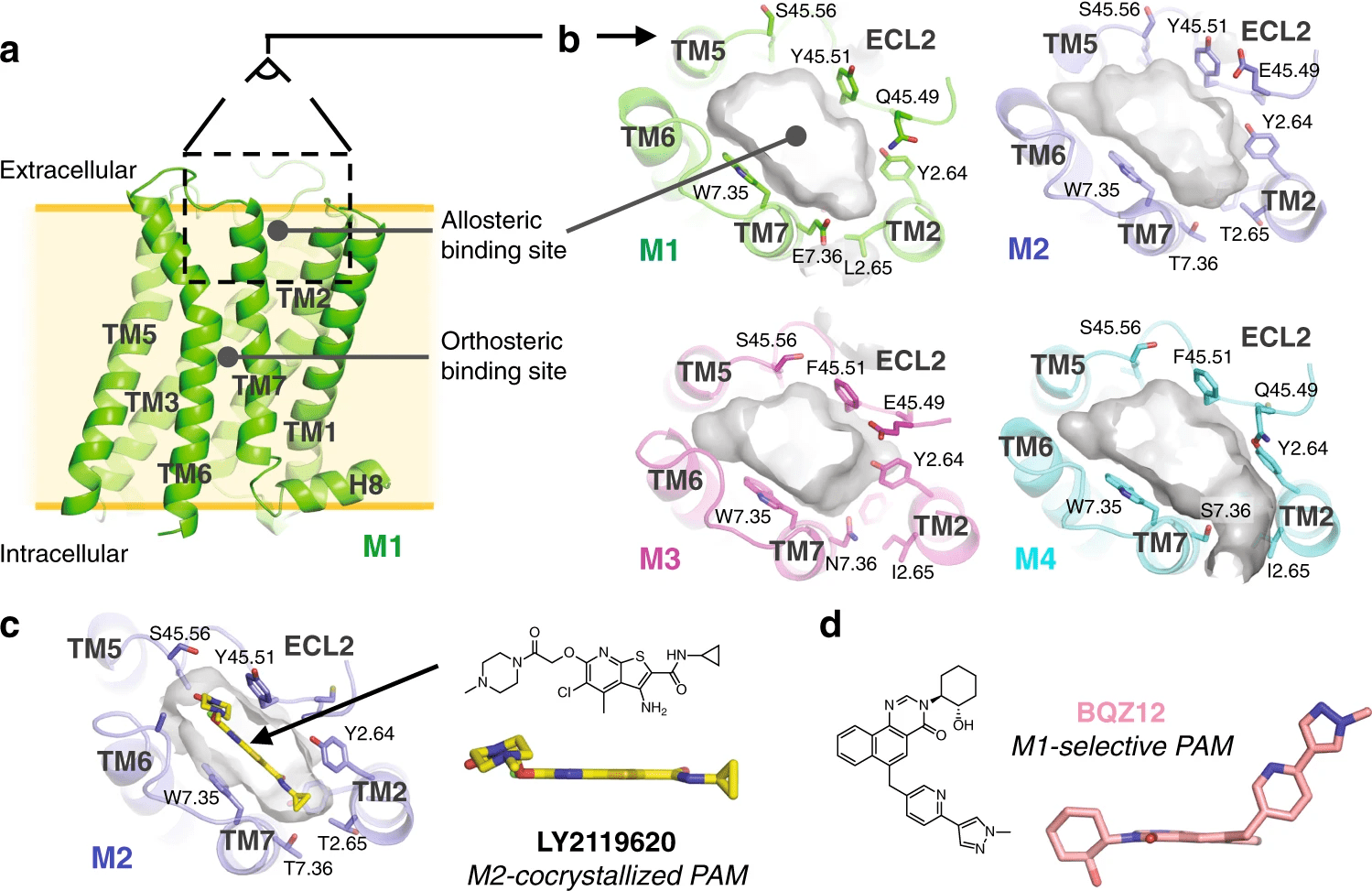
Detecting Cryptic Binding Sites
Cryptic sites are hidden in the unbound structure and require conformational changes to become apparent.
Strategies
- Use enhanced sampling MD methods like metadynamics.
- Apply pocket detection to multiple conformations.
Case Study: Identification of allosteric sites in Hsp90
Ligand pose optimization
After today, you should have a better understanding of
Accurate Docking Depends on Optimized Ligand Poses
- Precise ligand poses are crucial for reliable predictions of binding affinity and activity.
- Incorrect poses can lead to false negatives or positives, misguiding drug development efforts.
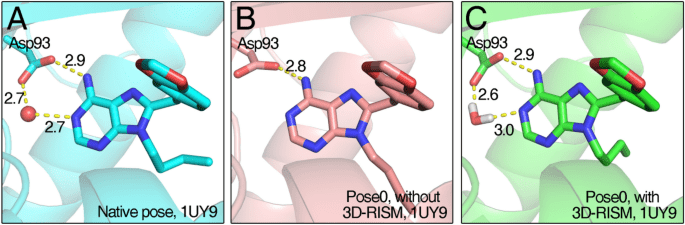
Fundamentals of Ligand Pose Optimization
-
Definition of Ligand Pose
- The specific orientation and conformation of a ligand within the binding site of a target protein.
-
Optimization Goal
- Identify the energetically most favorable pose that closely represents the true binding mode.
-
Key Components
- Orientation: Position and alignment within the binding pocket.
- Conformation: Internal geometry, including bond angles, lengths, and torsions.
Docking needs to generate diverse conformations
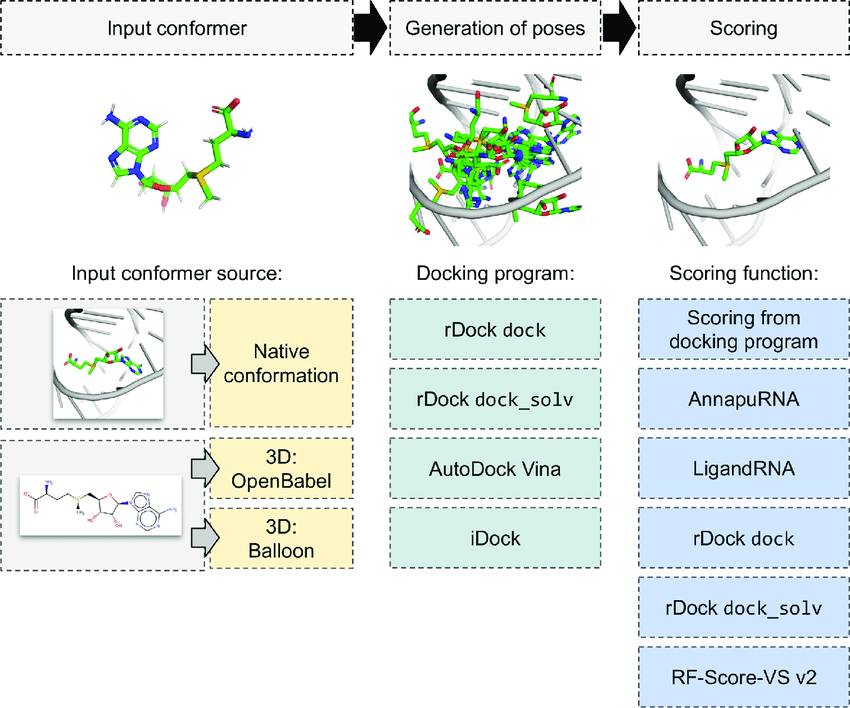
Search strategies
- Systematic
- Stochastic
- Empirical
- Machine learning
Systematic searches numerically iterate over all possible conformations

Identify important degrees of freedom
- Angles
- Dihedrals
Scan along each angle with a step size of a N degrees
Remove structures with high strain
Systematic searches are only possible for very small molecules
How many different conformations would we have in this molecule if we scanned only dihedrals every 45 degrees?

Systematic searches are only possible for very small molecules
8 dihedrals
1
2
3
4
5
6
7
8
8 angles
8 × 8 × 8 × 8 × 8 × 8 × 8 × 8 = 16,777,216
That's a lot of structures, and many of them will clash!
We almost never do a systematic search in practice without some precautions to combinatorics
Stochastic algorithms provide better balance of sampling and cost
Monte Carlo
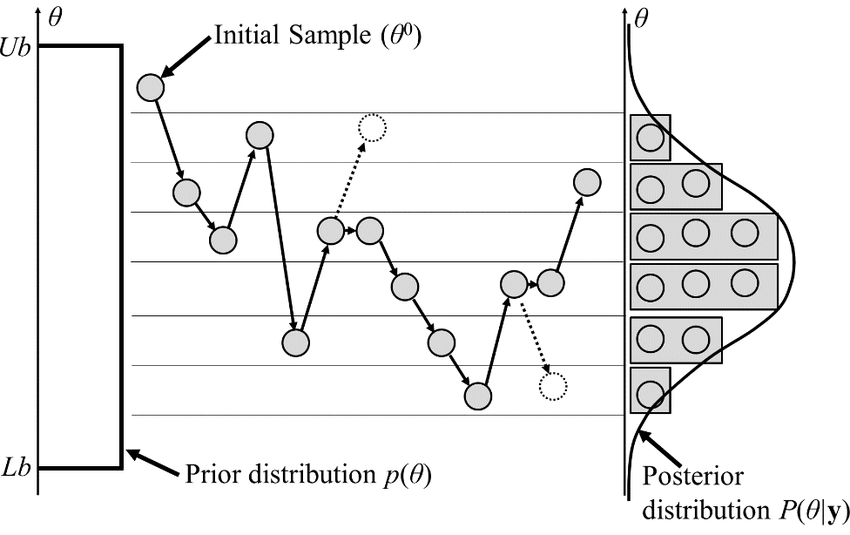
Steps:
- Generate conformation
- Compute energy change
- If energy change less than a random sample: make move
- Repeat
Allows us to sample efficiently
We also have conformer libraries
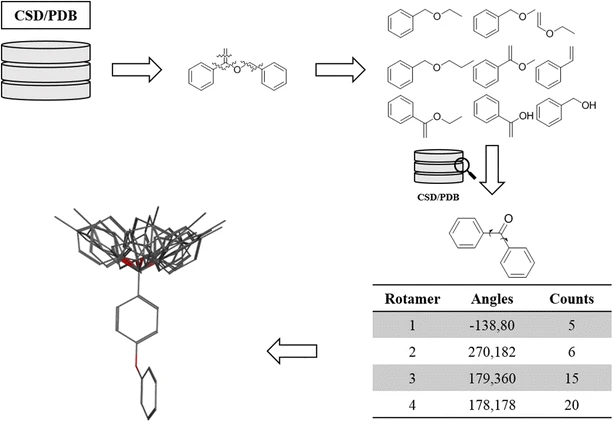
Use pre-generated libraries
Scoring functions as data-driven predictors
After today, you should have a better understanding of

Scoring function are parameterized models to estimate binding affinity after docking


Physics-based methods using force-field like methods
Recently, machine learning (e.g., graph neural networks) have been gaining traction
Interpretation of docking results
After today, you should have a better understanding of
Case study: MAP kinase ERK2
Overview of ERK2:
- Extracellular signal-regulated kinase 2 (ERK2), encoded by the MAPK1 gene, is a crucial component of the mitogen-activated protein kinase (MAPK) signaling pathway.
- ERK2 regulates vital cellular processes, including proliferation, differentiation, and survival.
Role in Disease:
- Aberrant ERK2 activity is implicated in various cancers, often due to mutations in upstream components like RAS and RAF, leading to uncontrolled cell growth.
- ERK2 is also involved in inflammatory diseases and neurodegenerative disorders.
AlphaFold 2 Prediction
Before the next class, you should
Lecture 17:
Docking and virtual screening
Today
Thursday
Lecture 18:
Ligand-based drug design
Evaluating and Scoring Binding Pockets
- Volume and Depth: Larger, deeper pockets may accommodate larger ligands.
- Hydrophobicity/Hydrophilicity: Influences ligand binding characteristics.
- Amino Acid Composition: Presence of key residues for interactions.
Challenges and Limitations
-
Protein Flexibility
- Static structures may miss important conformations.
-
Water Molecules
- Role of water in the binding site can complicate detection.
-
Computational Resources
- High-resolution methods may be computationally intensive.
-
False Positives/Negatives
- Overprediction of non-druggable sites or missing true sites.
Accurate and efficient binding predictions are essential
Objective: Directly predict binding affinity from protein and ligand structures with high accuracy and minimal computational resources.
Why Empirical Data Matters
- Experimental data provides a solid foundation for predictions.
- Enhances reliability over purely theoretical approaches.

Thermal shift assays
High-Quality Datasets Enable Better Predictions
-
Utilizing Crystallographic Structures
- Real protein conformations guide accurate docking.
- Reveal detailed information about binding sites and interactions.
-
Incorporating Experimental Affinities
- Adjust predictions based on known binding strengths.
- Calibrate computational methods using empirical measurements.
Empirical Methods Improve Docking Accuracy
-
Utilizing Crystallographic Structures
- Real protein conformations guide accurate docking.
- Reveal detailed information about binding sites and interactions.
-
Incorporating Experimental Affinities
- Adjust predictions based on known binding strengths.
- Calibrate computational methods using empirical measurements.
Empirical Data Reduces Computational Effort
-
Efficiency Gains
- Focus on experimentally validated binding modes.
- Limit the search space to plausible interactions.
-
Resource Optimization
- Achieve accurate results without extensive computations.
- Save time and computational power for other tasks.
Empirical Data Increases Confidence in Results
-
Validation of Predictions
- Compare computational outcomes with experimental data.
- Confirm the accuracy of docking models.
-
Enhanced Decision-Making
- Reliable predictions support better drug discovery choices.
- Reduce the risk of pursuing ineffective compounds.
Introduction to Data-Driven Approaches
Data-driven approaches leverage large datasets and machine learning algorithms to make predictions, identify patterns, and generate models without relying solely on physical or chemical principles.
These methods harness the power of available biological data to accelerate research and discovery processes.
Advantages of Data-Driven Approaches
- Speed and Efficiency: Capable of processing and analyzing large datasets rapidly, facilitating quicker decision-making.
- Scalability: Easily scalable to handle growing amounts of data without a significant increase in computational resources.
- Flexibility: Adaptable to various types of data and applications, from genomics to drug discovery.
- Cost-Effectiveness: Reduces the need for expensive experiments by predicting outcomes computationally.