Role of DNA methylation in the genomic rearrangements in P. tetraurelia




PhD Defense DELEVOYE Guillaume
09/06/2022
Supervisor : Dr MEYER Eric
Jury members : Dr DUHARCOURT Sandra, Dr CHEN Chunlong, Dr DURET Laurent
Qu'est-ce que l'ADN ?



Le noyau
des cellules
est rempli
d'ADN
Où se trouve l'ADN ?
A quoi ressemble l'ADN ?
ATCGATGCGGATTCGATCATGCTAGCTGATCGATCGGAAGCTTGACTAGTCGATCGATCGATCGATCGATCGATCTTCTATATATATGCGCGTAGCTAGCTAGCTAGCTATATATGCATAGAGAGCTCGATCGCGCTATCTCCTCTGATCGATCGATCGGGATCGATCGGATCGATGCATTAGGATCGATCGGT
....

A quoi ça sert ?
Entre autres, c'est un livre de recettes de protéines



Certaines recettes... S'autorépliquent
La protéine qui correspond à un bout d'ADN peut parfois copier-coller, ou couper-coller ce bout d'ADN ailleurs
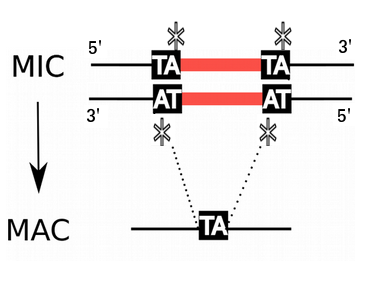
P. tetraurelia: Genomic architecture
Unicellular eucaryote with 3 nuclei:
-
2xMIC nuclei (2n)
- Germline nucleus
-
Contains: TE & IES
- No transcription outside meiosis
-
1xMAC nucleus (up to 800n)
- Somatic nucleus
- Amplified and "fixed" version of the MIC
- Free from TE and IES
- Transcriptionnally active
DNA ratio: 1 MIC for 200 MAC
+ DIfficult to purify the MIC DNA
Profiling the IESs
- Non-coding
- Excised after sexual processes
- Remnant of TE ?
- Most recent IES are very short (< 27bp), and are the majority of IESs
45.000 Unique sequences
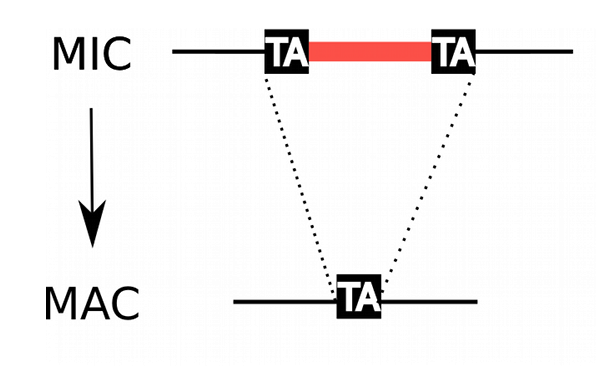
How are IESs recognized ?
(1/2)
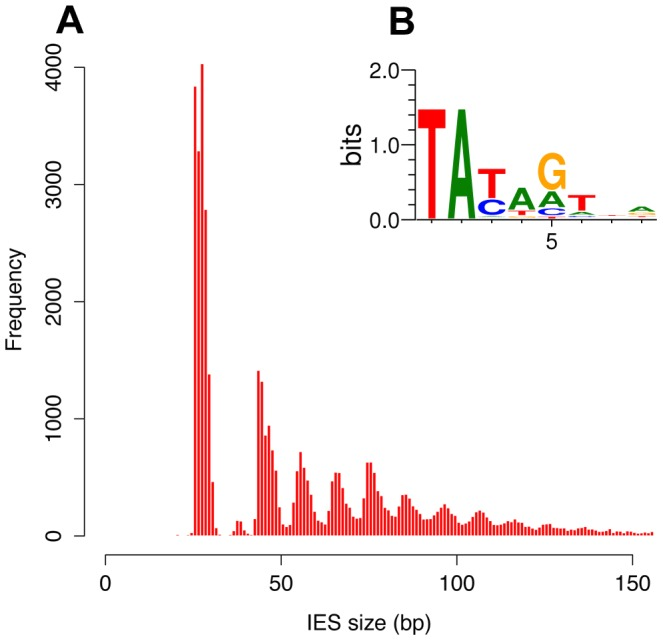
- 100% TA-Bounded
-
Weak consensus TAYAG
- Degenerated TC1-Mariner TE insertion site
- Not sufficient
- Periodic size distribution
30% of IESs only are small-ncRNA dependant (shown by DICER-like2-3 silencing)
- What about the majority remaining ?
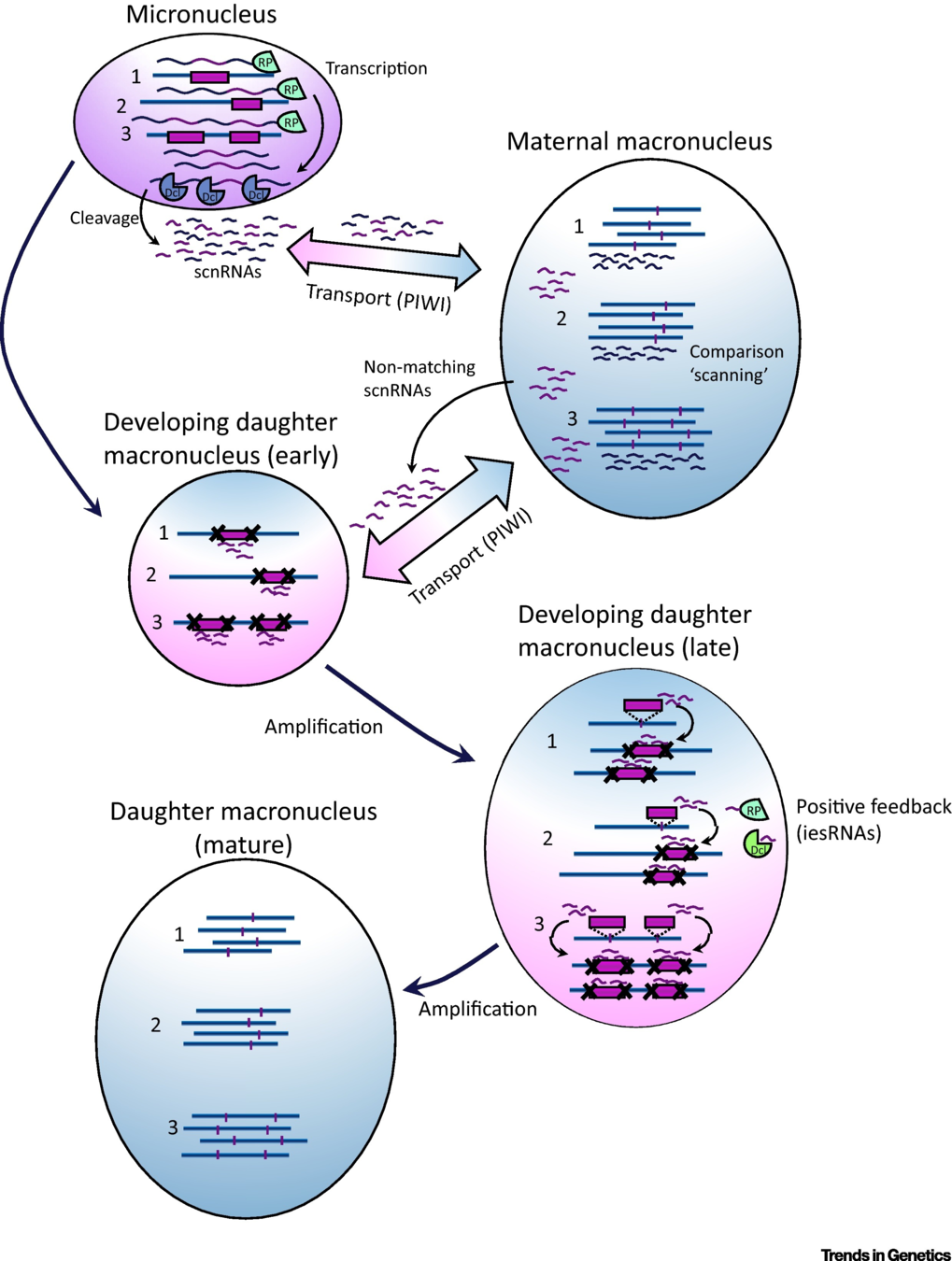
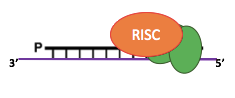
How are IES recognized ? (2/2)
The sc-RNA pathway
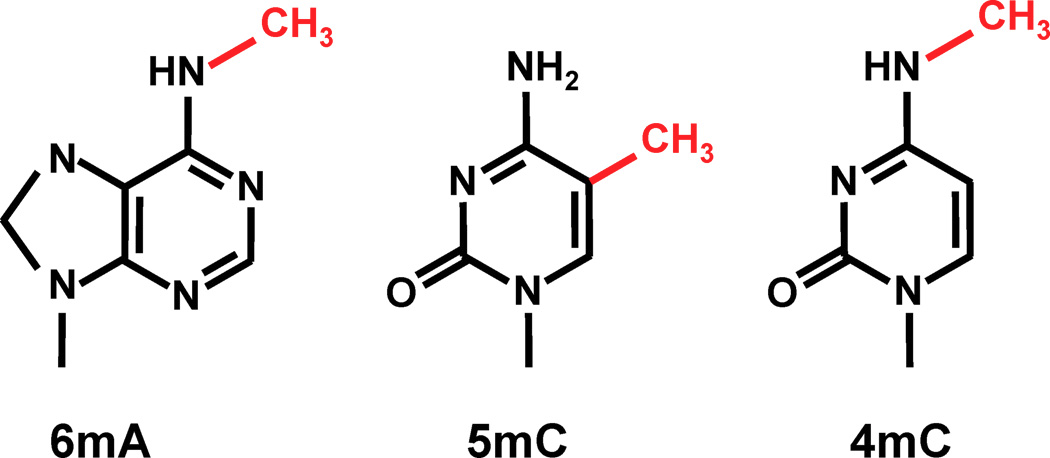
The DNA methylation hypothesis
6mA likely to be abundant in Paramecium:
-
Suspected 2.5% in the MAC of P. aurelia by Cummings et Al (1975)
-
Also documented 6mA in the MIC
-
-
-
Detected methylation by SMRT in the MAC in our lab (unpublished data)
-
Detection by SMRT in the MAC by Sandra Duharcourt's lab (unpublished)
- Detection in Oxytrichia by L. Landweber et Al (2019)
- no 5mC, 4mC in the MAC a priori

2) Crosstalks in the new forming MAC
1) Constant pattern in the MIC
Transcient ?
Methylase candidates
Candidates:
- 7 "DPPW motif IV" genes
- NM4, NM9, NM10
- Another family: MT1a, MT1b, MT2
2) PacBio sequencing with short inserts | 6mA ++
Also at hand:
- Vegetative WT cells (HTVEG)
- Vegetative cells with a silencing of a control gene (MAB)
- Cells that undergoe autogamy (HT2 - 2 hour , HT6 - 6 hour)
1) Grouped silencings by sequence homology
PacBio
TL;DR overview
PacBio SMRT principle
Methylation analysis needs local coverage> 25X
- IPD are captured
-
Compared
- either to WGA
- or Trained model (ML - "in-sillico control") - SImulation from context
$$log(ipdRatio)= log(\frac{MeanIPD_{experience}}{Model})$$
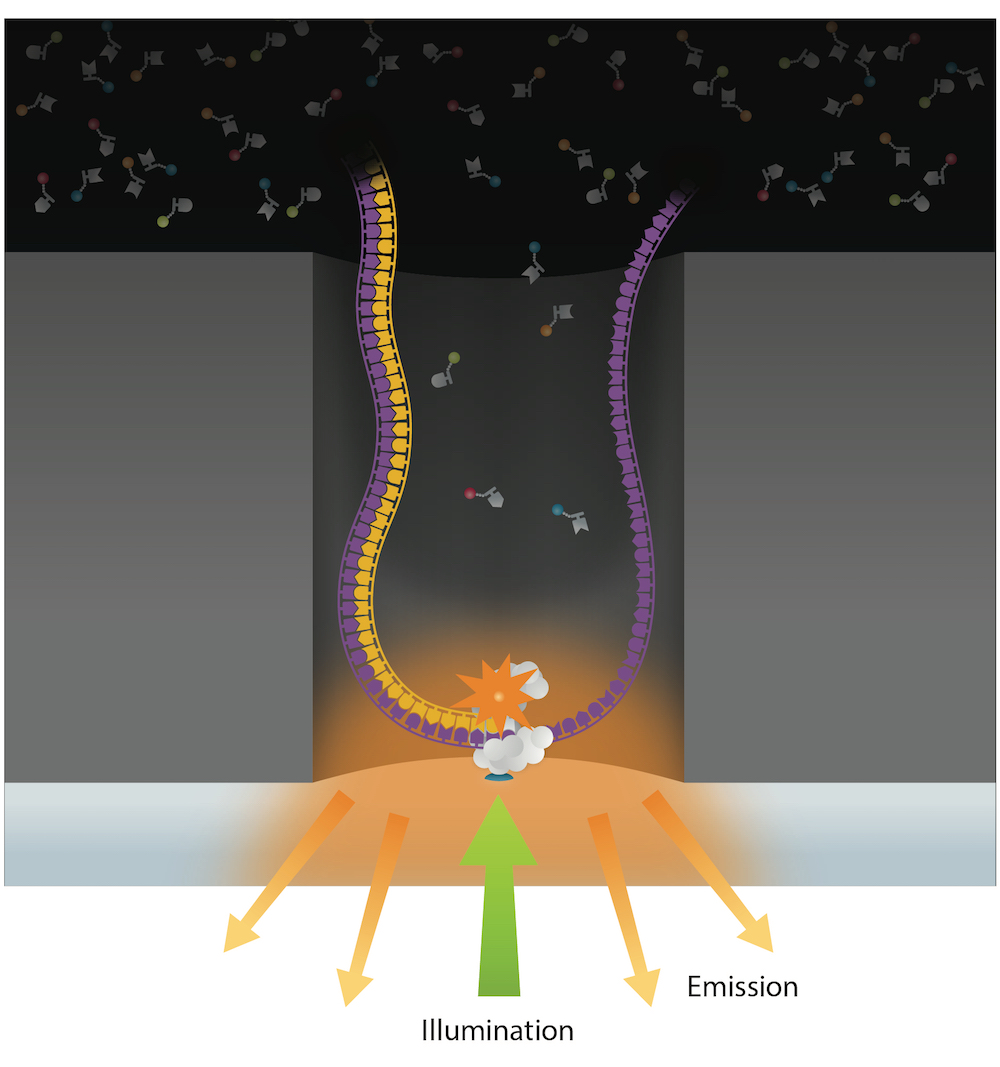
Reads up to 80 kbp
- We use the isn-sillico model
- Nobody does
PacBio SMRT principle (2)
- Strategy 1: Long inserts (long reads)
- Ideal for assembly of long repeated sequences
-
Poor resolution for DNA methylation analysis
-
Strategy 2 : Short inserts (long reads)
- Much higher resolution for DNA methylation analysis
Step 1: Consensus
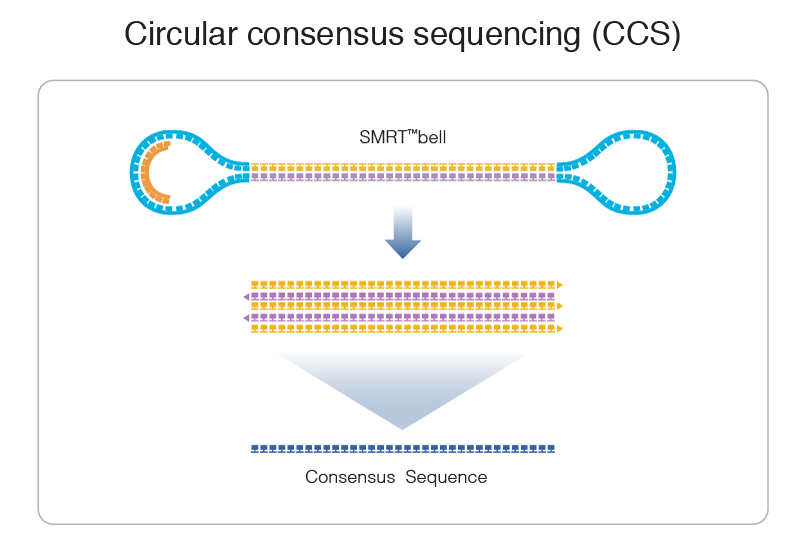
99% accuracy
Max
75% accuracy
Because our inserts are circular and shorts, we can make CCS of high accuracy despite a 15% error-rate
Step 2: Sorting
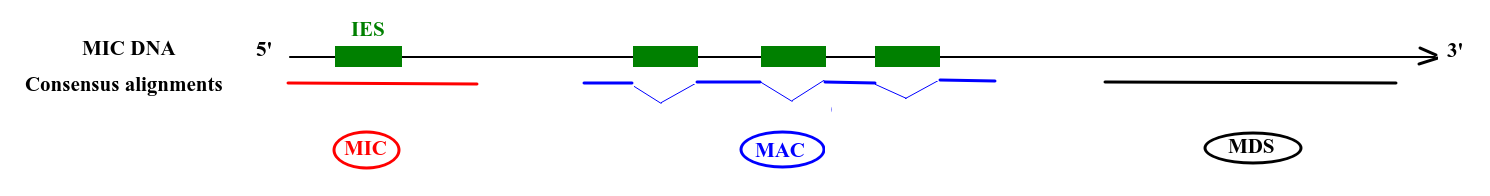
Deduced origin
MIC DNA
Alignment of consensus
TA
TA
TA
Step 3 : DNA methylation analysis
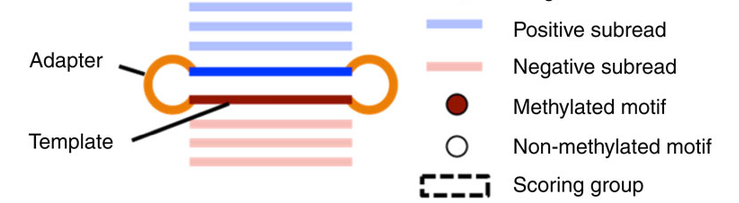
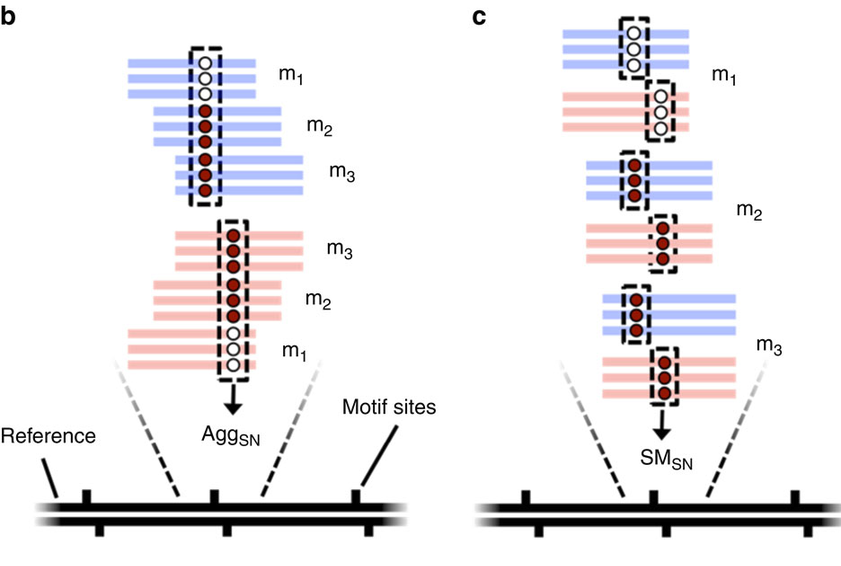
- Single-molecule
- Single-nucleotide resolution
- Independant yet pairable analysis on both strands
Some more details:
the random sampling approach
Deduced origin
MIC DNA
Alignment of consensus
Only a few remaining: ~ 10 to ~200 sequences
100% should carry a methylation pattern
-
1 out of 200 comes from the MIC
-
1/6 of MIC inserts will carry an IES
-
1/2 of IESs are just wrongly retained in the MAC
- 30% of the remnants are scanRNA dependant
Final categories
- MDS
- = "MAC" for 99% of sequences
- MAC_IES
- "true" MAC_IES (never seen retained)
- other MAC_IES (sometimes retained)
- MIC
- Other MIC specific (TE, repeated sequences)
- MAC (TA Junction)
- Overlap a TA junction of excision
- rDNA
- mtDNA
-
Other:
- Contaminants
- Alternative excision boundaries ("LOWID")
- low identity consensus ("Trash")
"genome"
Total Paramecium
Total sequencing
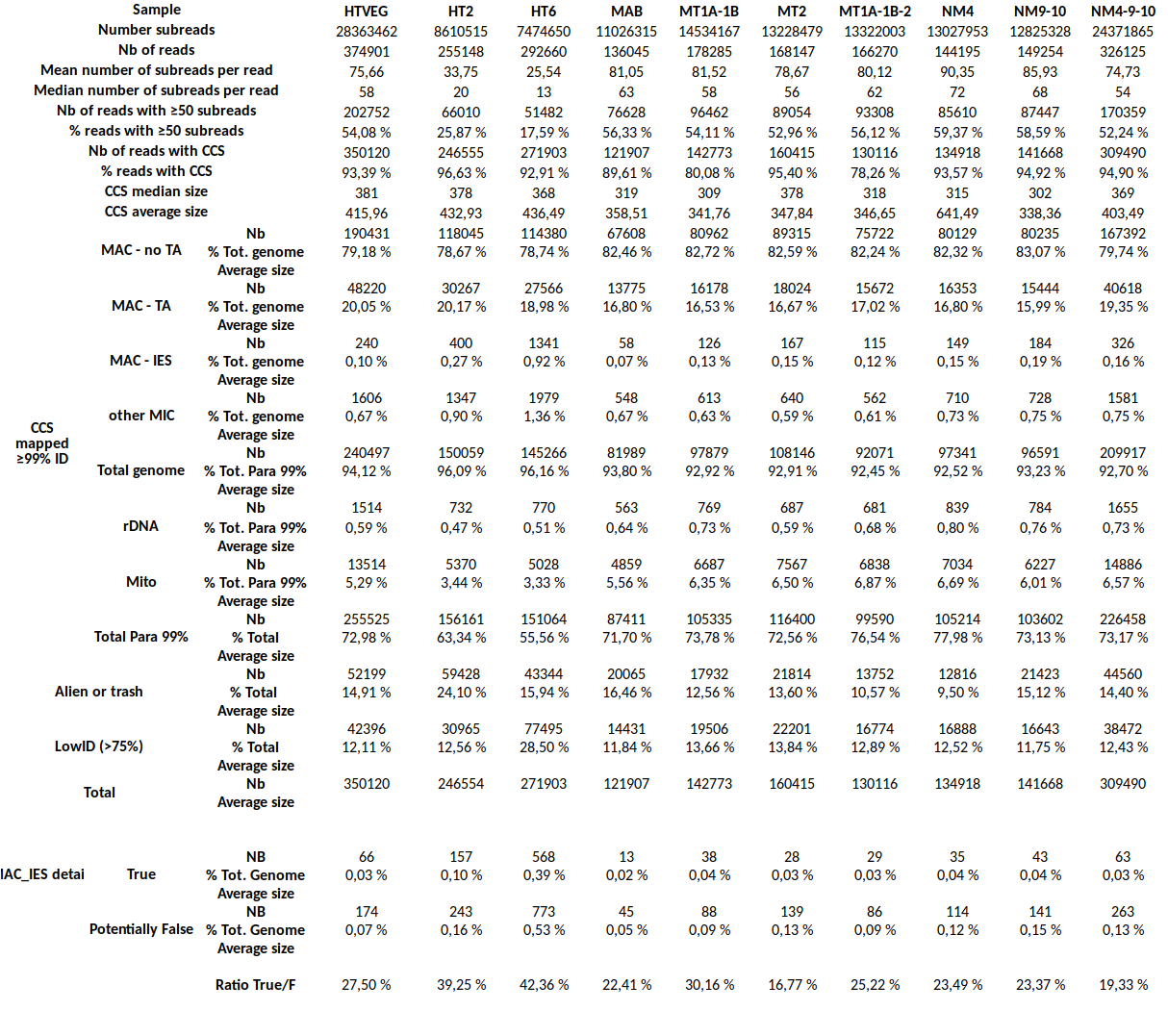
Little bug to be corrected
30%
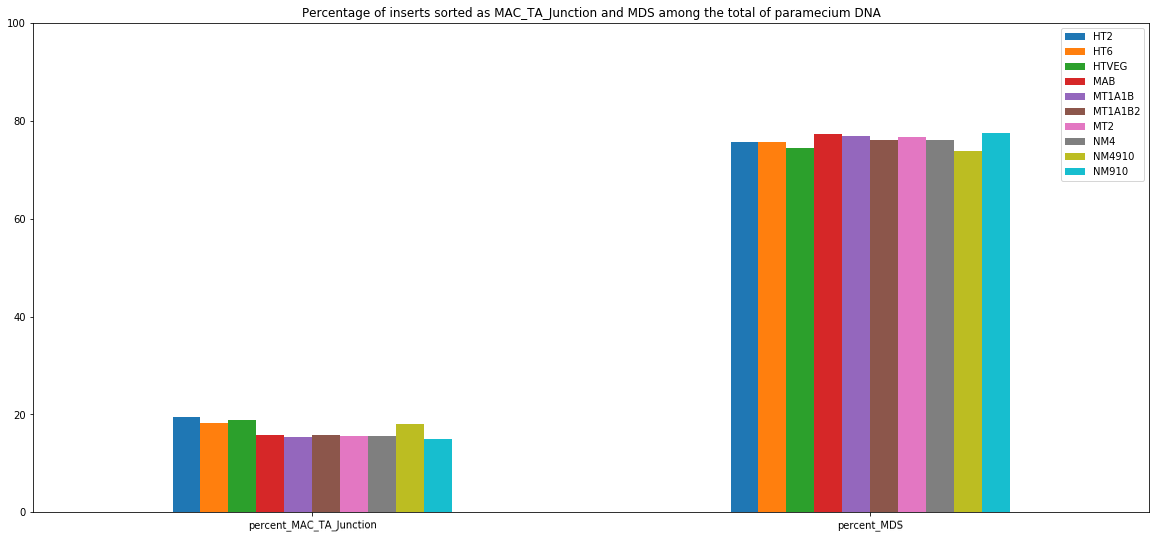
MDS and MAC_TA_Junction represent a vast majority
MIC specific and IES are as rare as expected
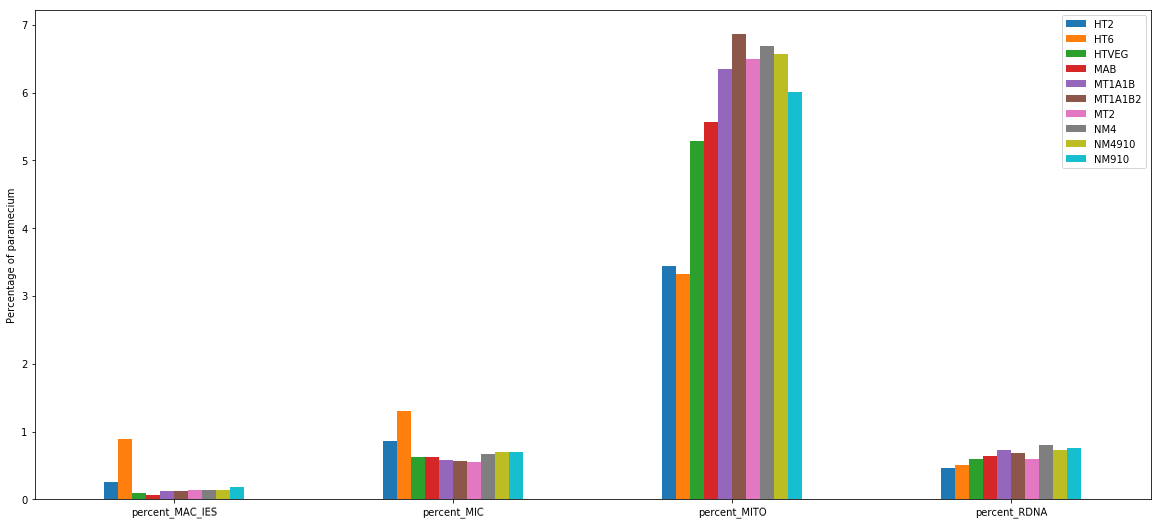
+ Applying the retention filter divides the number of "MAC_IES" approximately by 50%
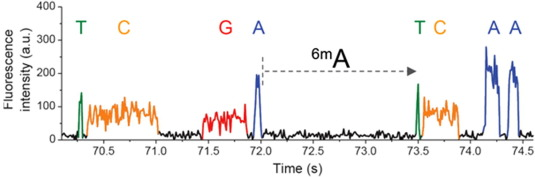
Detecting m6A in details
Detecting m6A optimally
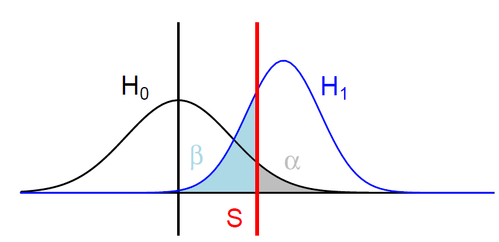
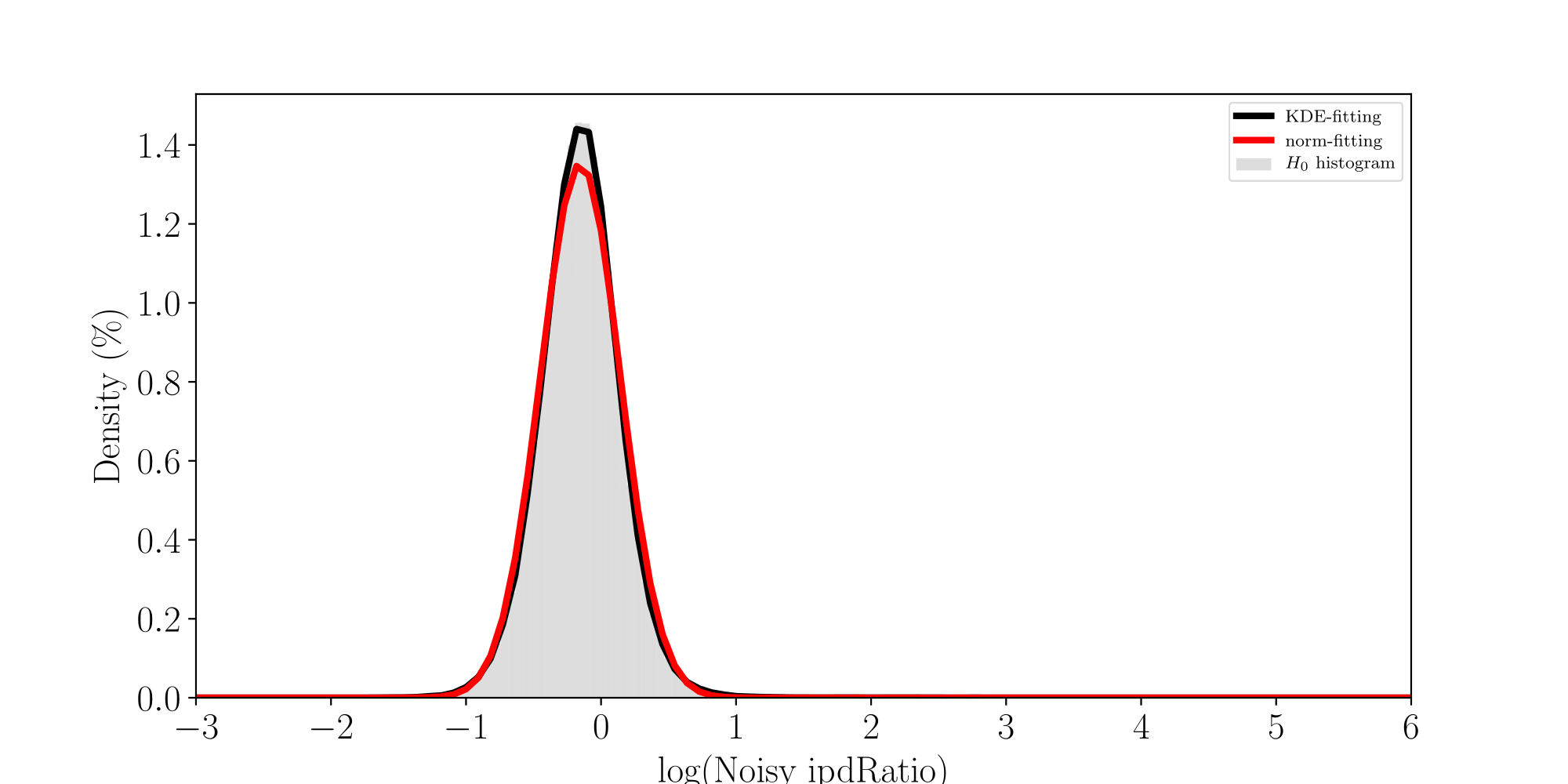
H0: ipdRatio of umethylated-Adenines
H1: ipdRatio of 6mA
S: Threshold on the pvalue
--> Specificity
--> Sensitivity
$$\alpha$$
$$1 - \beta $$
Adenine
6mA
ln(ipdRatio) ~ N(0,1)
log(ipdRatio)
A pvalue is just the likelihood that log(ipdRatio) is in the tail of H0 when H0 is true
Using E.coli as ground truth
E.coli:
- Feeds paramecium: contaminants+++
- Nearly 100% symetrically-methylated with m6A
- GATC
- EcoK
- Few others
- Depends a lot of the strain
- Outside of GATC and EcoK: very low levels of m6A
I investigated the PacBio's output on it's GATC & EcoK VS other sites
How log(ipdRatios) look in E.coli
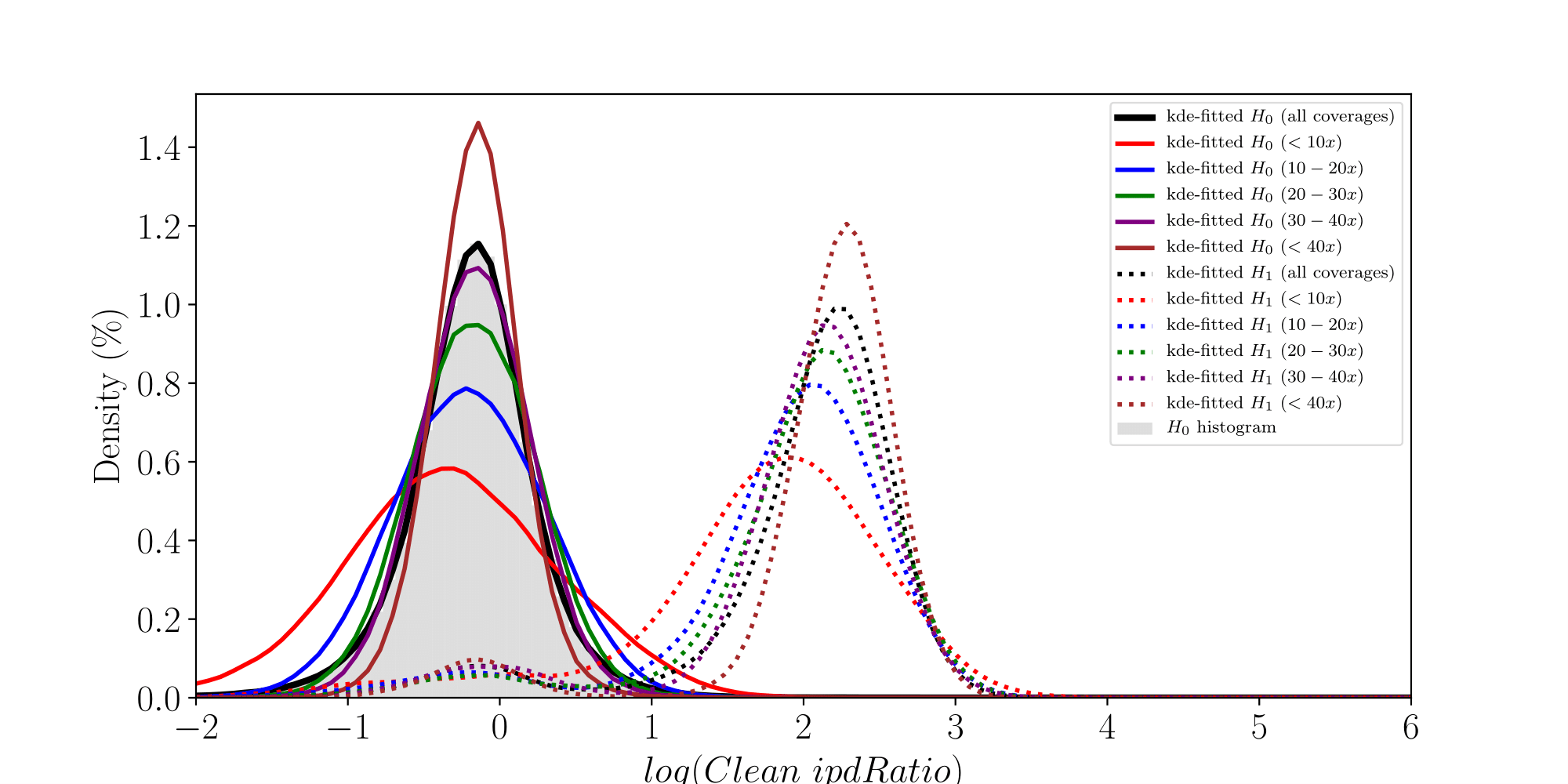
Separability raises with coverage, which is expected
PacBio pvalues do have some biological meaning
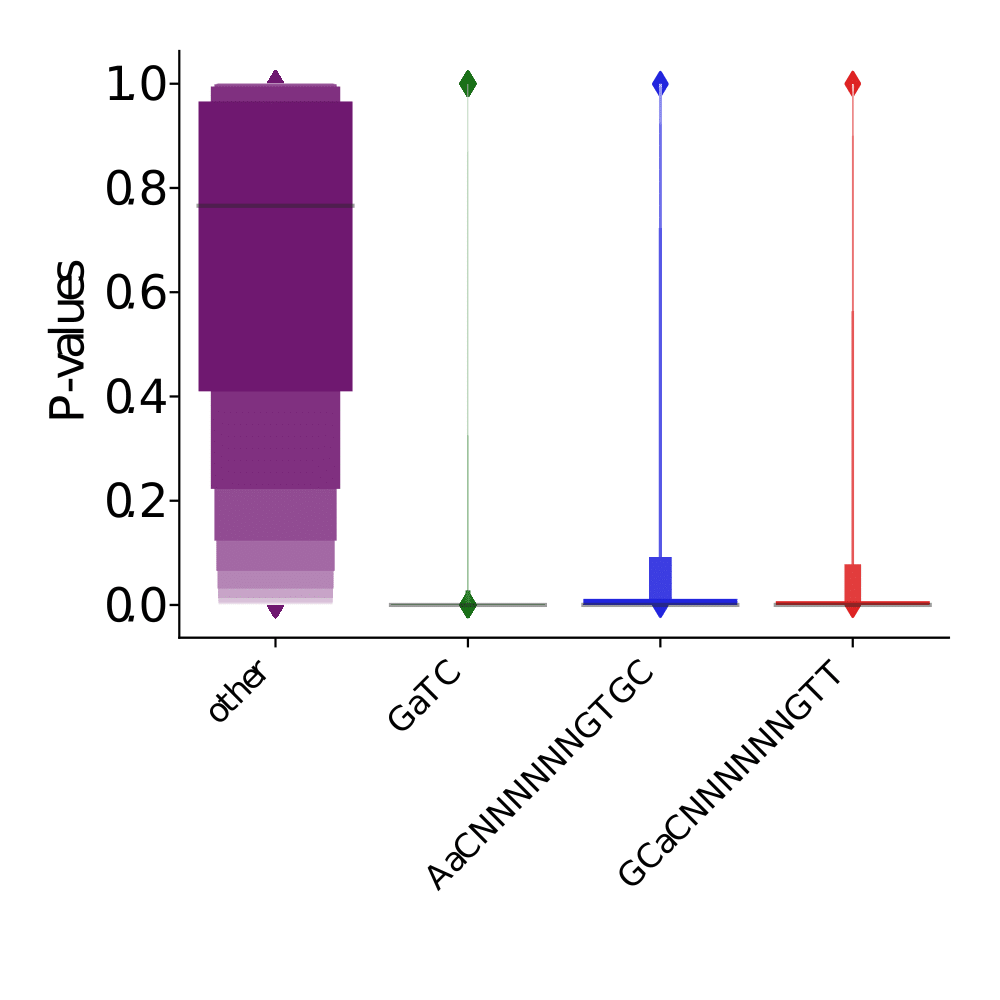
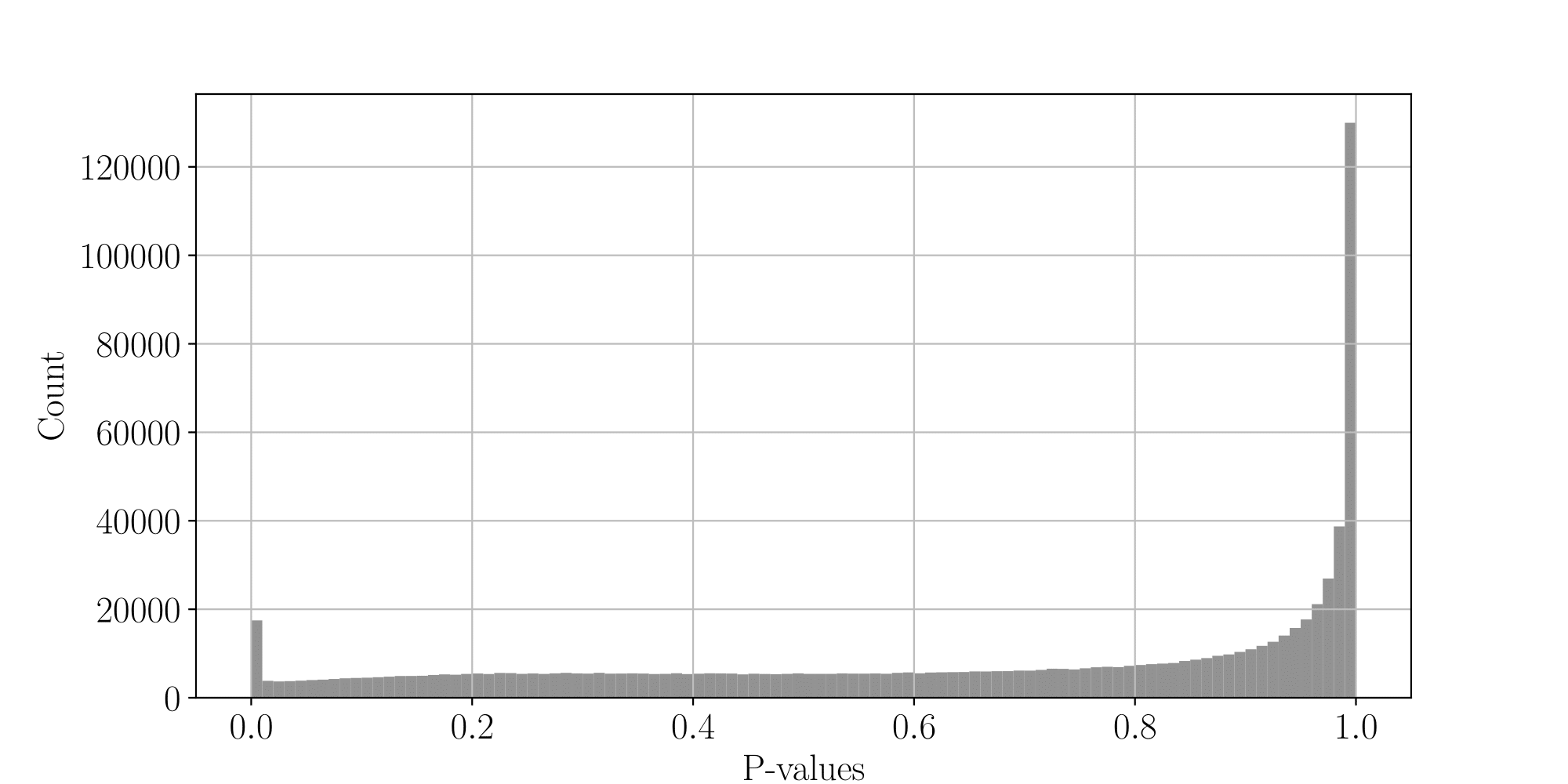
PacBio pvalues do have a meaning...
...but are not ideally distributed
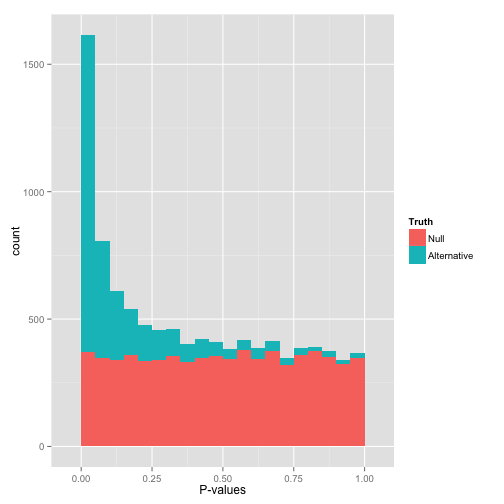
Ideal pvalues
--> Allows magic !
PacBio's
- Allow estimation of
- Allows optimal, adaptative FDR control
$$\pi_{0}, \pi_{1}$$
- Just don't
-
Obvious point n°1:
- log(ipdRatio < 1) -> -inf
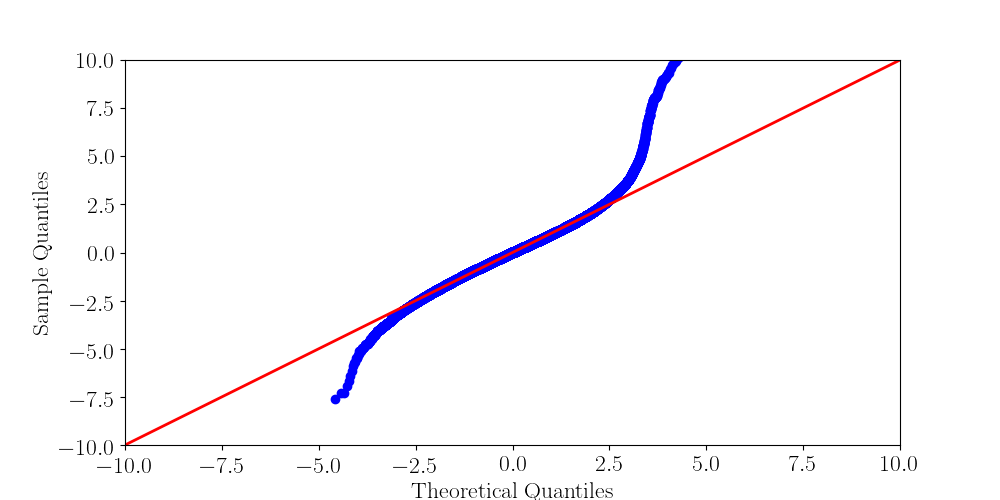
Normality assumptions under H0 are broken under high coverages
- The higher the coverage, the worse (here, >40x)
-
Will cause bell-shaped pvalues under null
- Hidden phenomena on the previous curve
Normality assumption matters
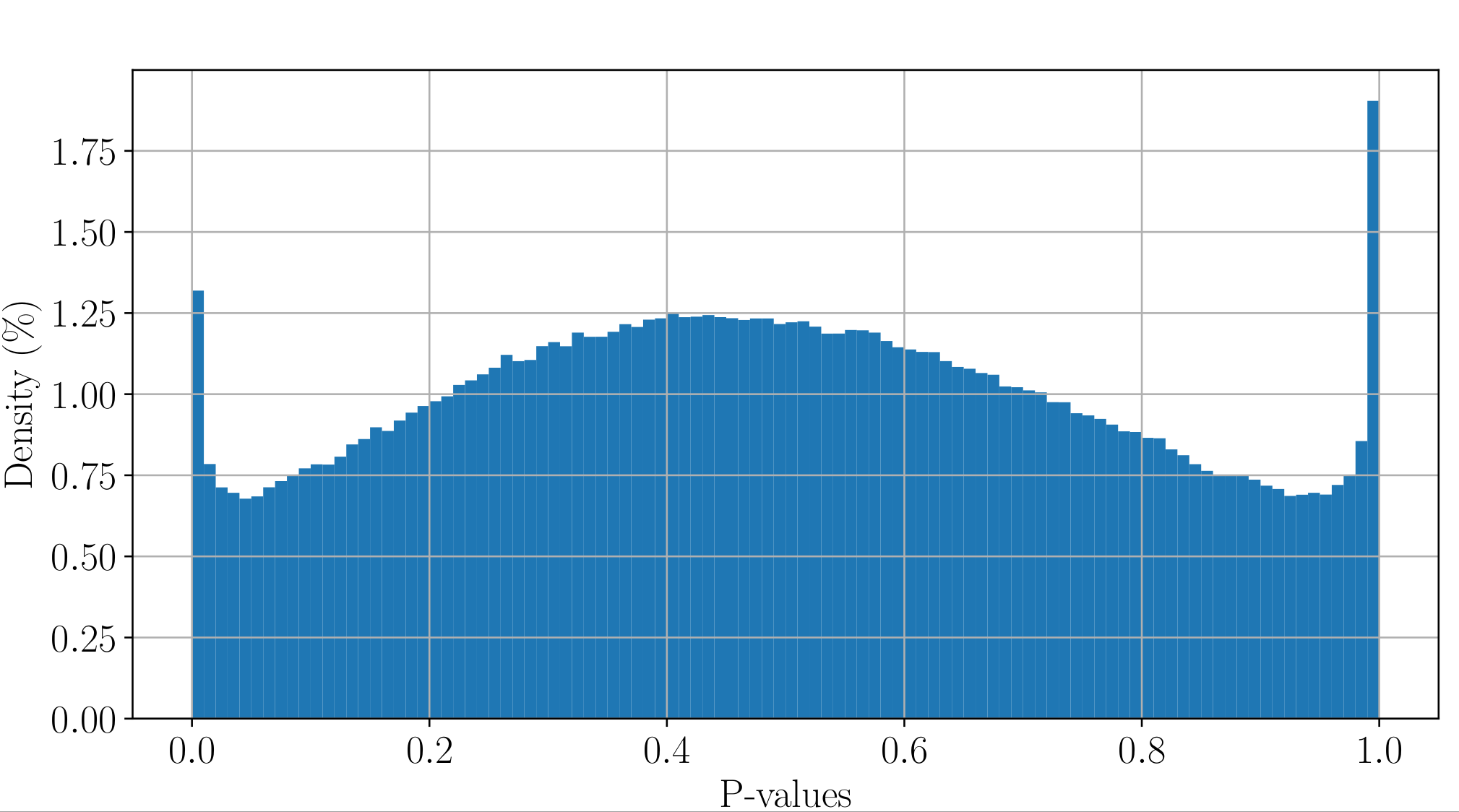
All Adenines' pvalues [E.coli] coverage > 40X
Long story short
PacBio's pvalues:
- Are biologically relevant
- We can build a reliable ad-hoc system with them
- Are produced by a linear combinations of > 150 different coverages
- Which forbids the usual statistical treatments
- Are somehow broken on a coverage-dependant manner
- Which forbids a simple fix for point 2
- We can't use the classical statistical treatments directly on pvalues
Other PBio's scores for m6A
For n6mA, PacBio produces:
-
A modification score --> Slowing of the polymerase
- pvalue against H0 only (the one we presented earlier)
-
An identification score --> Kinetic signature of a modification
-
loglikelihood between H0 and H1 (H1 = Other modifications or secondary peaks)
-
loglikelihood between H0 and H1 (H1 = Other modifications or secondary peaks)
They are PHRED-transformed p-values of two different statistical tests, that rely on the mean of the IPDs
PHRED scores
The scores (Qv) are PHRED-transformed p-values
Typical covscore plot
Modification score / coverage
Using flat threshold on modification score = Hudge lack of power
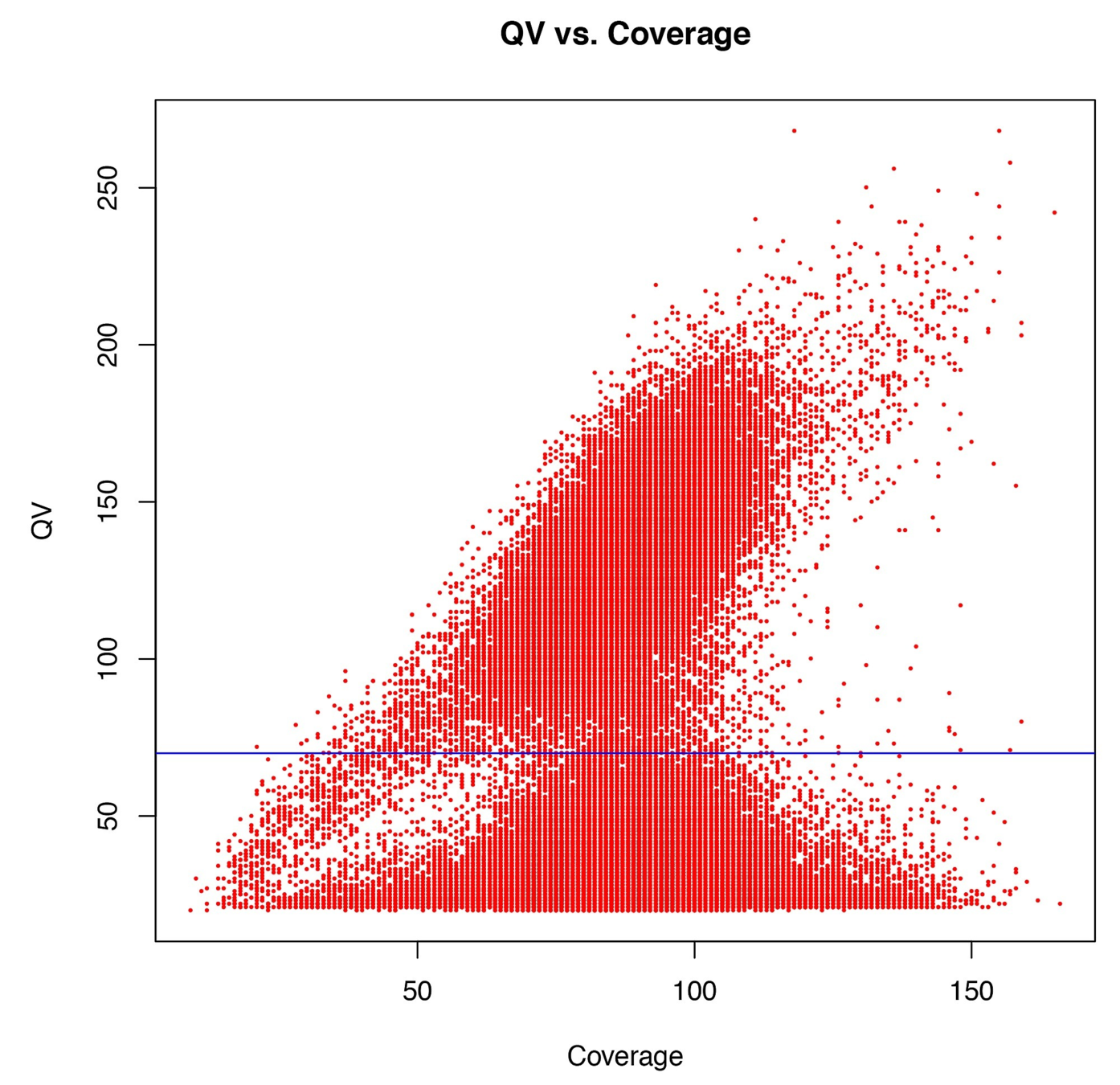
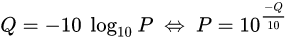
Solution : A coverage-dependant threshold on the scores
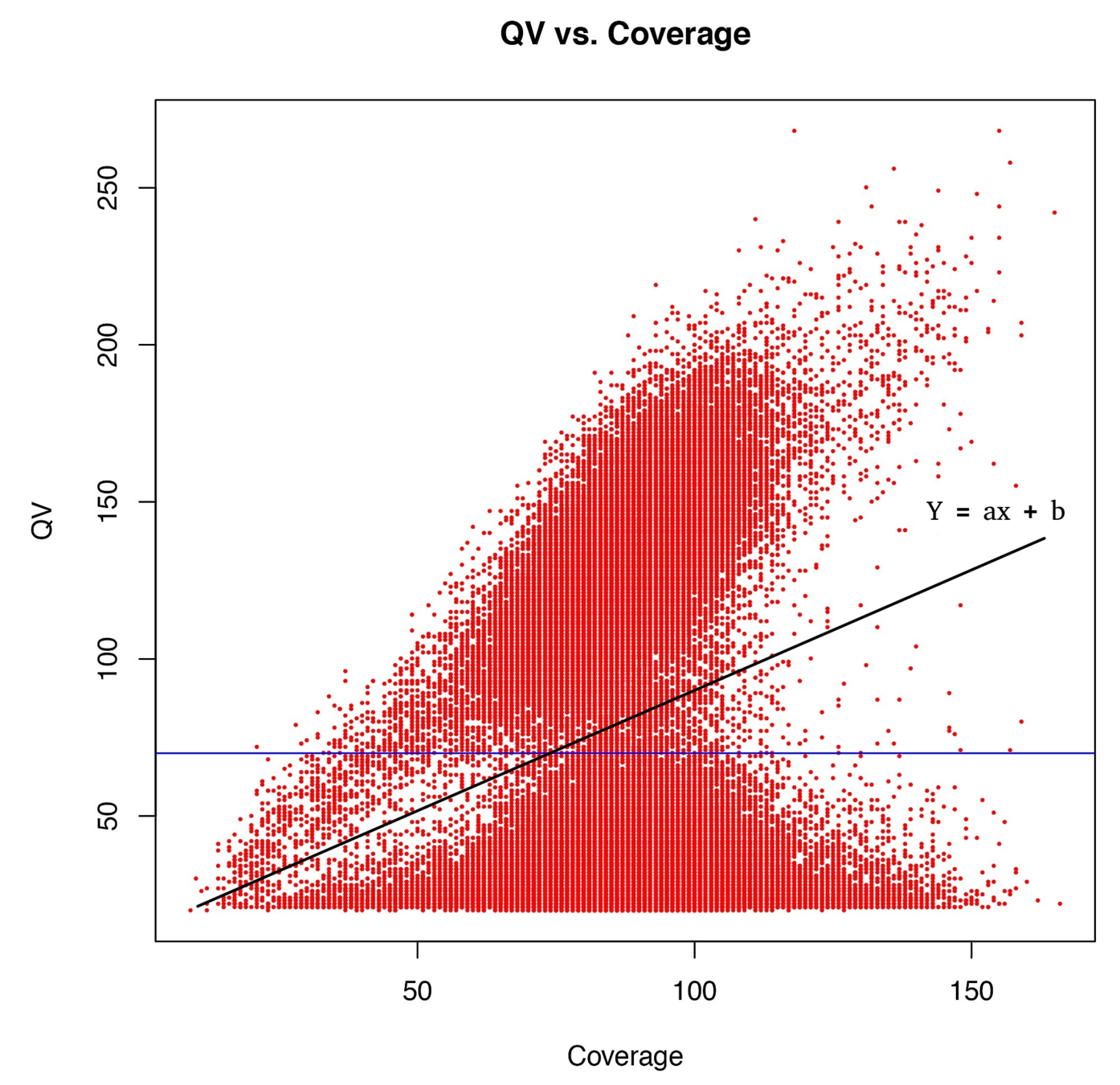
From now on
"positive detection"
=
score > linear thershold
(only >25X considered)
Benchmarking
How good (or bad) is our method ?
$$Se = P(D^+|M) ~ 92\%$$
$$Sp = P(D^-|NM) ~ 99.8\%$$
Starting from sufficient coverages (~20X to ~30X), Se and Sp don't depend on the coverage anymore
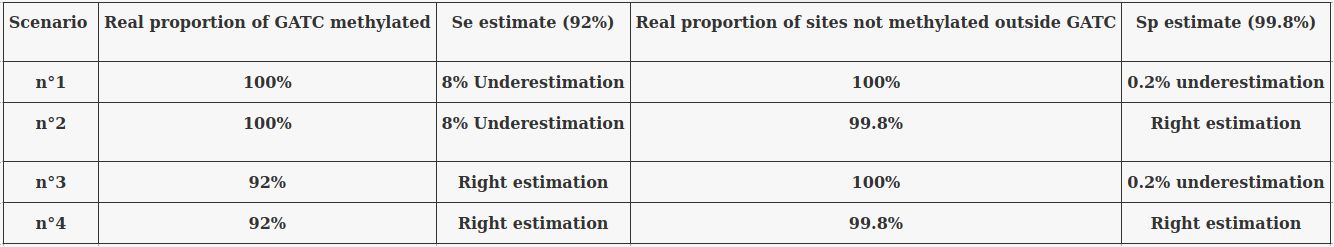
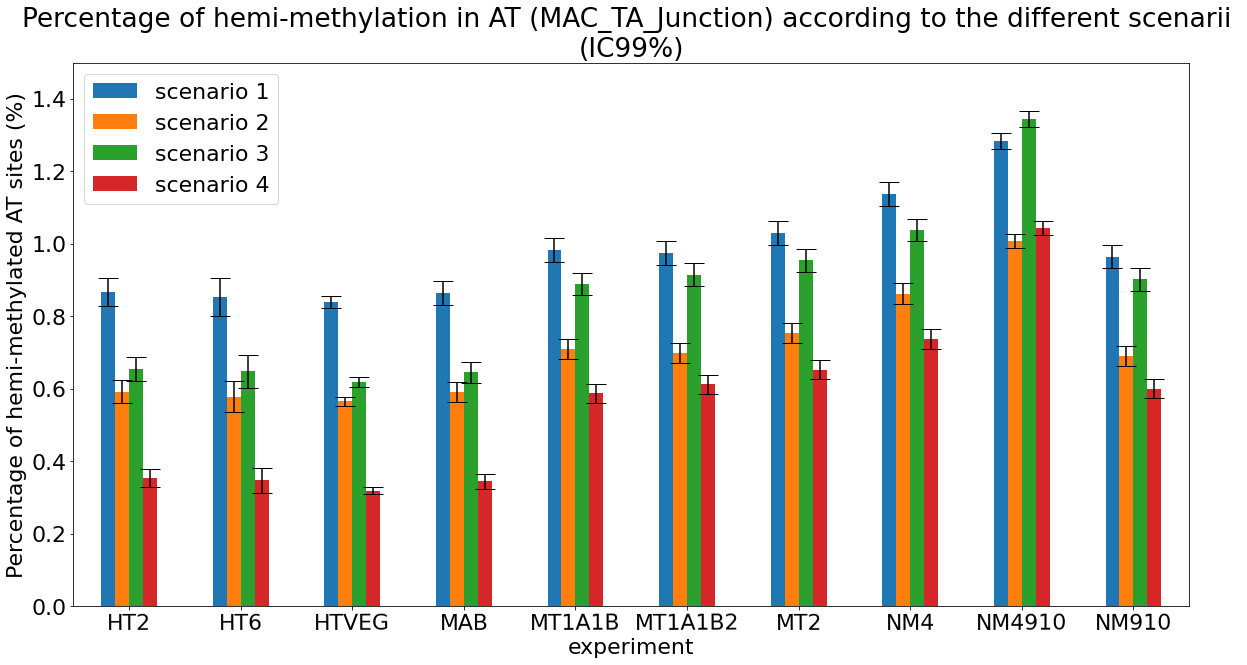
The 4 scenarii for Se and Sp
- We don't care about Se and Sp
- We care about the fact that eventual mis-estimations of them doesn't really change anything
Finding unbiased estimators for and FDR
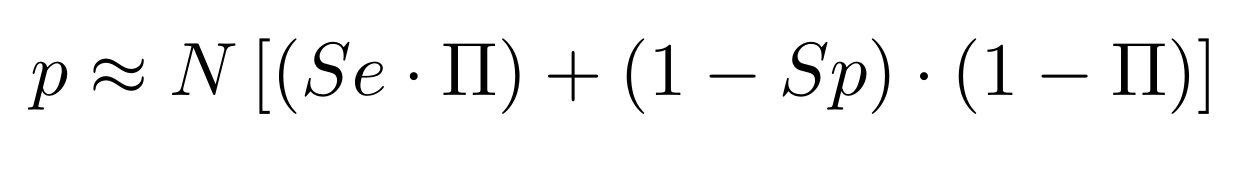
If p number of positive detections among N tests:
p = FP + TP
$$\pi$$
So,
Which means
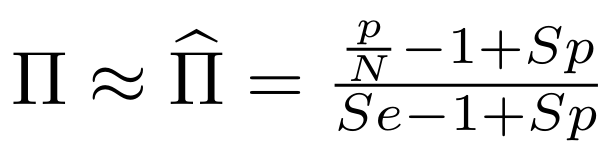
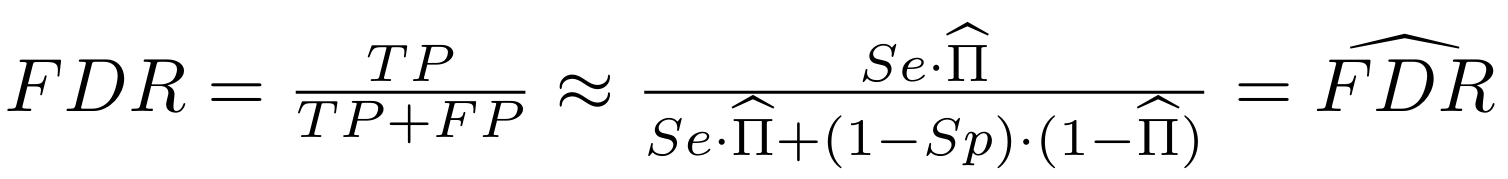
And:
What it gives in Paramecium
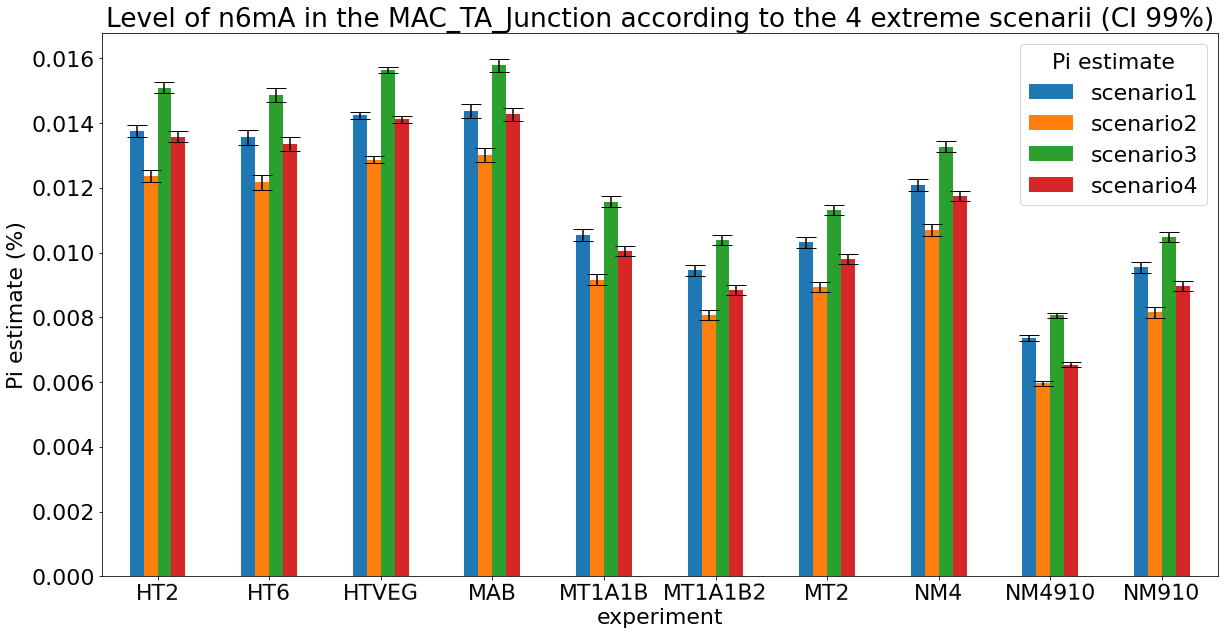
Debiased levels of m6A in the MAC_TA inserts
Details in MAC HTVEG
-
~95% of the methylation locates in AT dinucleotides in the MAC
-
True in any condition
-
-
75% of the methylation in an AT dinucleotide is actually symetrically modified, independantly from being in the MAC or the MIC
Kept in MIC and MAC
All conditions
MAC_TA outside of AT sites
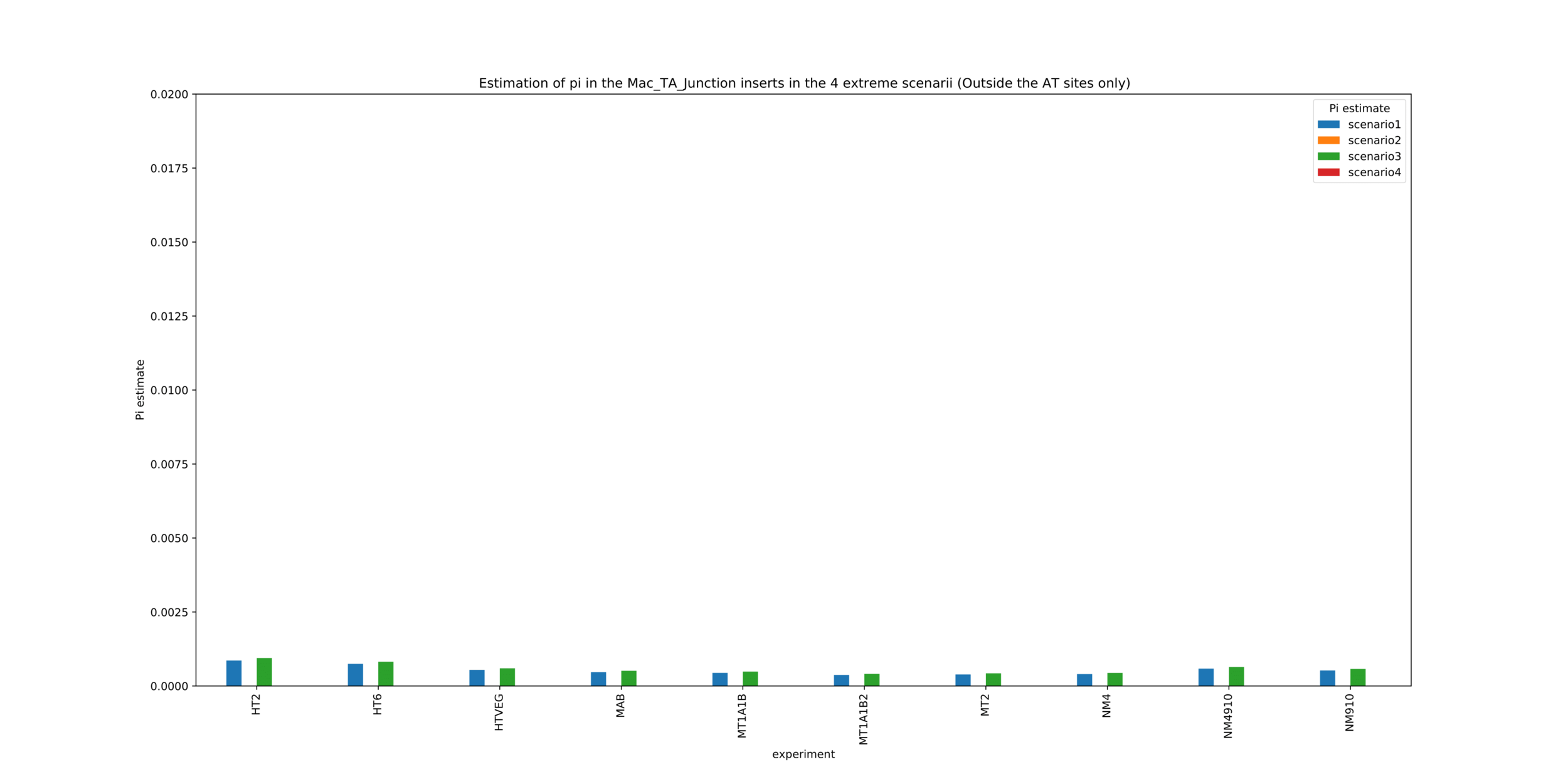
Detections outside AT sites are likely to be at least partly true positives
Present in all samples
Never erased
Conclusion: We are either in scenario 1 or 3
(Sp largely underestimated)
Was expected but confirmed
Methodological development to correct hemi-methylation detection (1/2)
Let FD1 and FD2 be resp:
- Fraction of hemi-methylated AT sites
-
Fraction of sym-methylated AT sites
PZ0, PZ1, PZ2: unbiased estimators of non, hémi, symetrically methylated AT sites
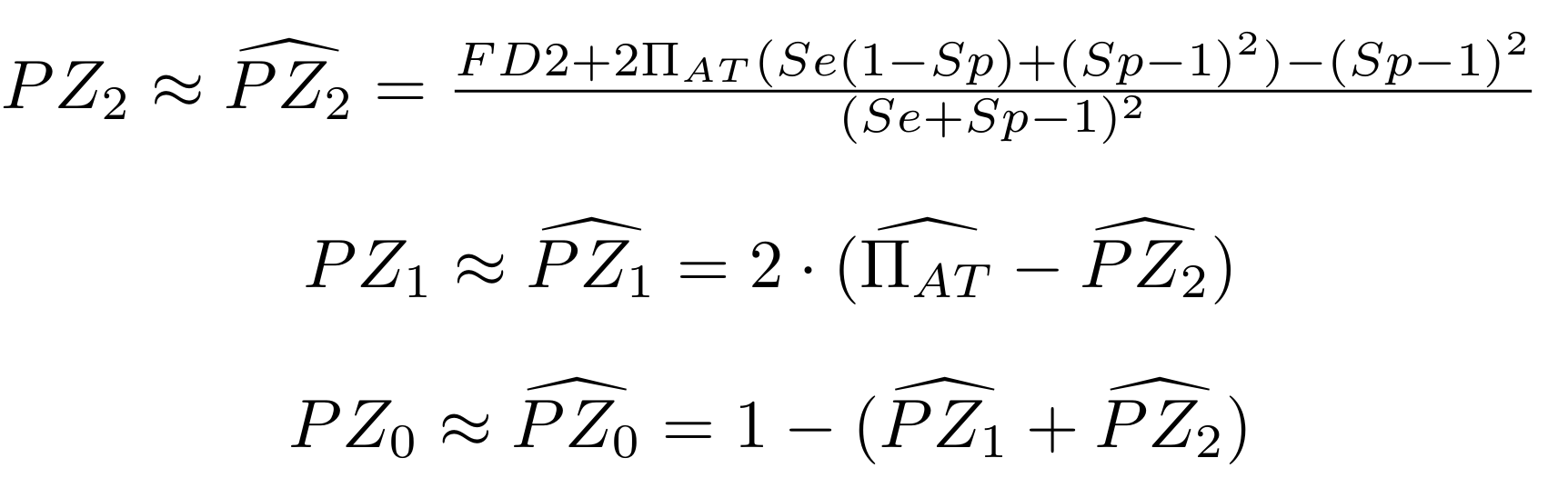
Then:
With
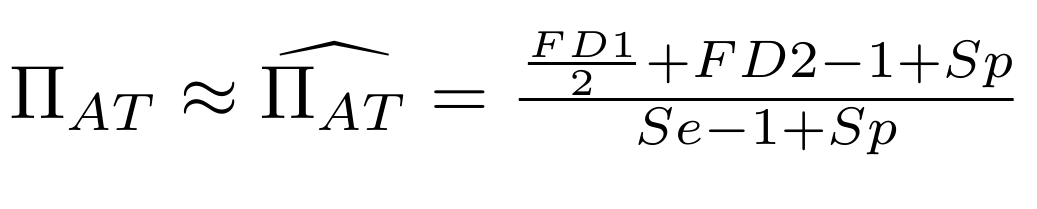
Methodological development to correct hemi-methylation detection (2/2)
We can also find the number of hemi-methylated sites being detected as such, and the proportion of sites detected as hemi-methylated that are really hemi-methylated. This is possible because we now approximately know PZ0, PZ1 and PZ2, and P(D|Z) is easy to determine:

Then, P(Z|D) can be determined through Bayes theorem using P(D|Z), P(Z) and P(D) (which are all known)
P(Z=1|D=1)
is our case of interest
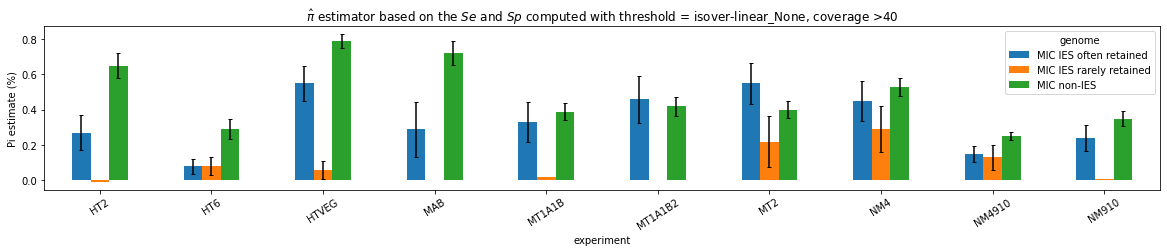
What it gives in Paramecium
Debiased Hemi-methylation in MAC_TA
n6mA in the MIC (preliminary)
Pure MIC sequences: Cannot yet be trusted (error in the pipeline)
Coverage >= 40X made us lose too many materials, should restart with >=20X
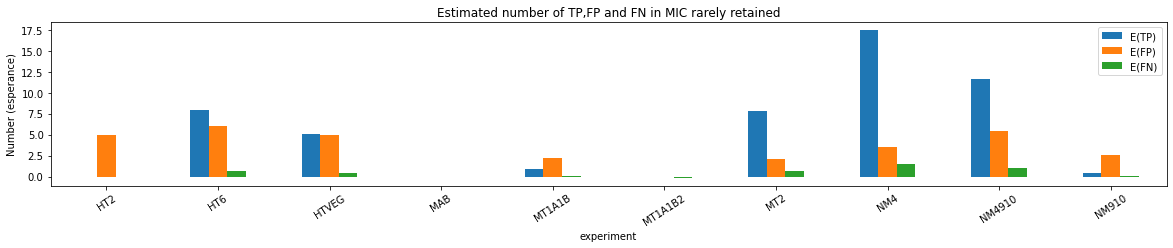
n6mA in the MAC_IES rarely retained ("TRUE MAC IES")

HTVEG

MT2
--> Some molecules carry all the detections, in sym-A*T
Very likely to be sequences comming from the MAC to my opinion
At first look, seems like the same in all samples
Conclusion
- Lots of sweat spent on methods : Now we start the cool things
-
In the MAC
- Quantification in the MAC is well-characterized in all the samples
- Our 6 genes are implied in the bulk of the MAC m6A
- MT and NM families: Functional analogs of DNMT1 ?
- Lots of questions raised by the MAC methylation: TSS, IES Junctions, nucleosome positionning...
-
In the MAC IES
- Very preliminary analysis between TRUE mac IES and other MAC IES tends to show that all detections could come from accidental retention in the MAC, and the MIC could actually carry 0% m6A or very lower levels than 1%. Really to premature to be sure
- HT2 and HT6 are still potentially full of surprises
- TE in the MIC: No idea yet
- It's just a question of time now ! Which we asked (prolongation LABEX memolife)

Sorry for the headaches !! Thanks for your time :)
2.1 Retroingineering
The capping of IPDs
-
modelPrediction is the predicted IPD value by the model in a given context of nucleotides at this position
-
globalIPD is the mean of all the IPD values of the read.
-
localIPD represents all IPDs that have been mapped at a given position in the genome, including those from other sequences
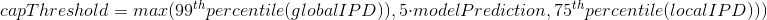
Conclusion on the capping
- Isn't coded as advertized by PacBio
- The way it's implemented for AggSN is problematic and doesn't really make sense
- Paradoxally, it should be more relevant for our approach than for the default one
- We expect no methylation to be undetected due to the capping
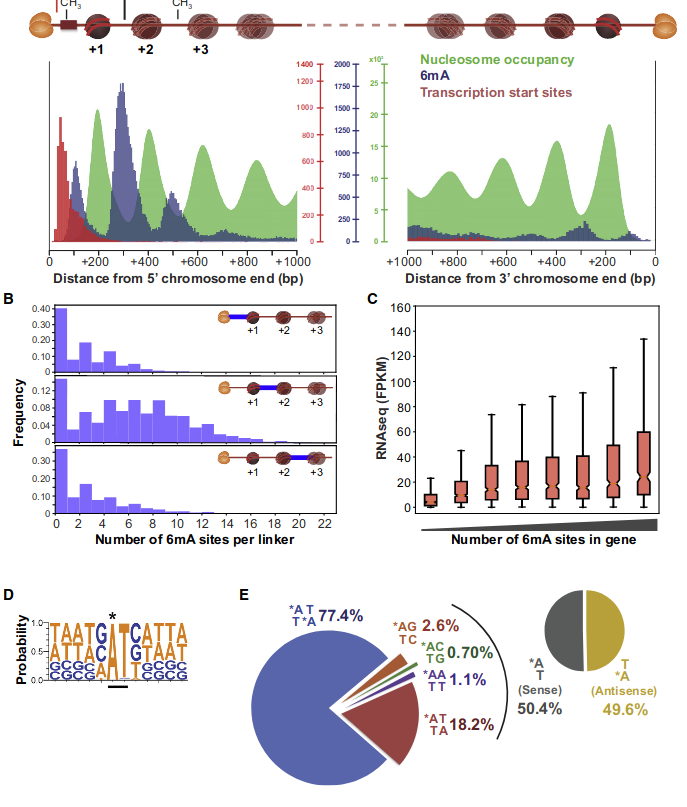
Laura landwebehr 2020
Oxytrichia trifallax
p values (A)
p-values (A score >20)
Out GATC score < 20
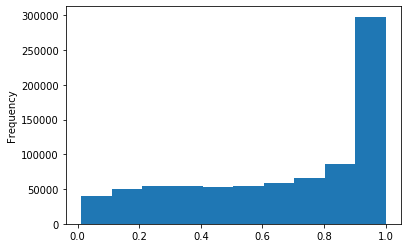
Out GATC
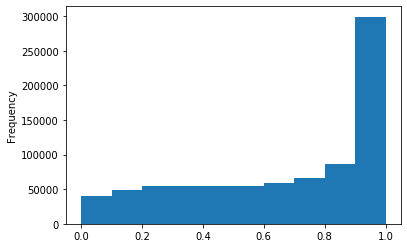
ipdRatio score 20
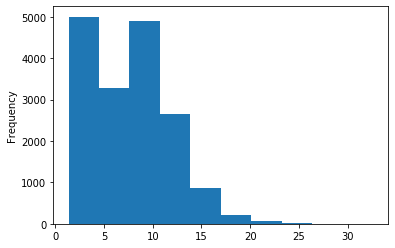
ipdRatio idv20/score20
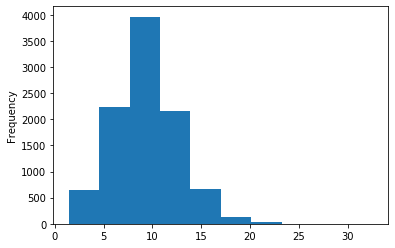
A outAT score 20 isQv20 (812 seq)
A outAT score20 idQv20 + Strong BH correction (176 seq)
ipdRatio out GATC before filtering BH vs after (qv20/idqv20)
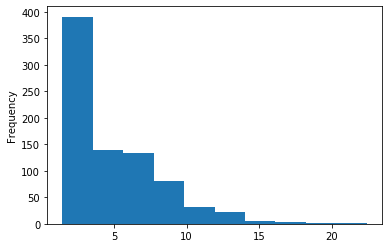
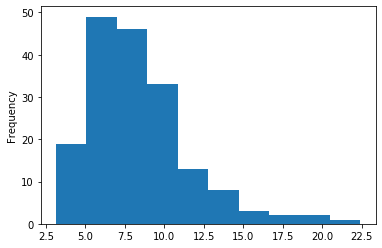
In GATC
