-
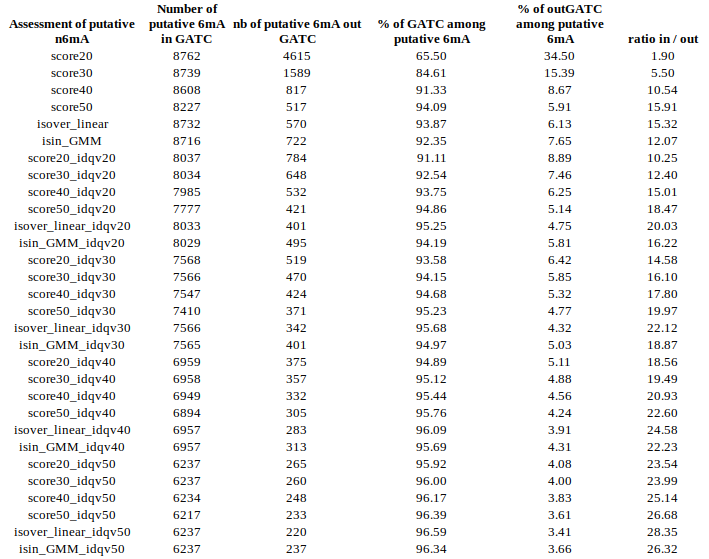
Advances on the MIC's n6mA
-
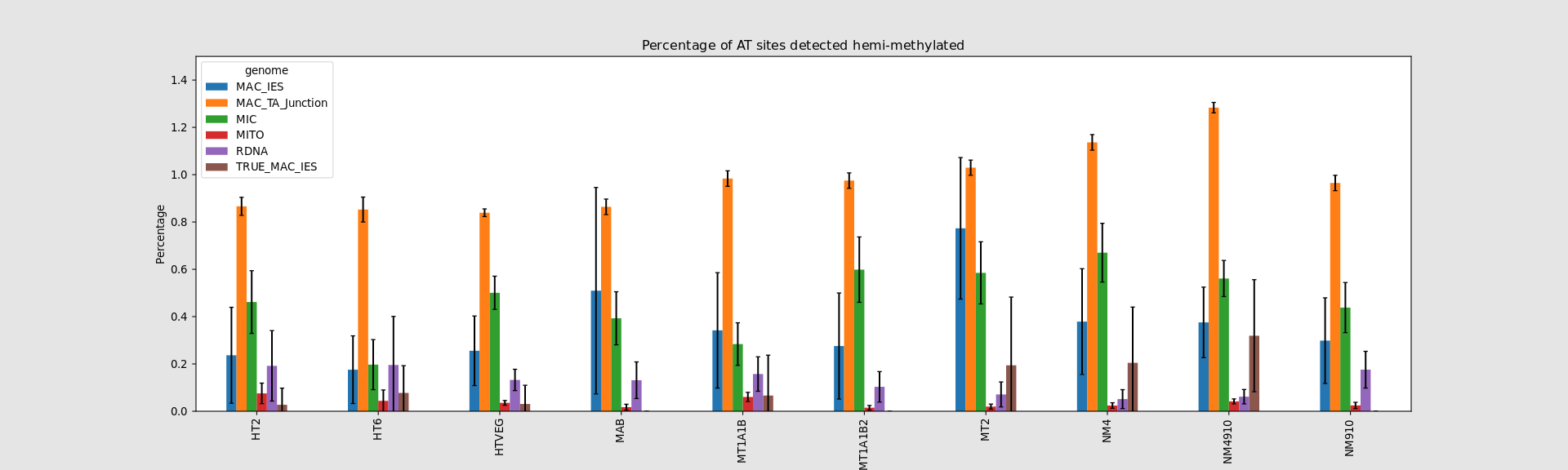
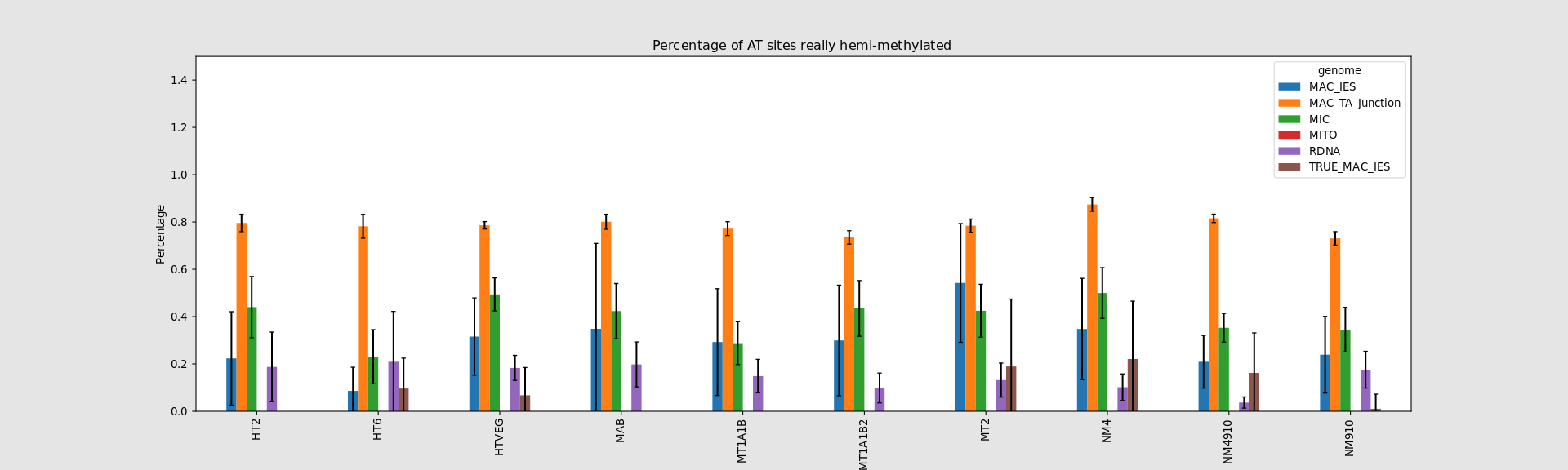
Hemi-methylated AT sites in the MAC
PhD Project
Biggest question: How does the cell recognize the Scan-RNA independant IES ?
2 independant hypothesis:
- Some motifs or specific nucleotide composition
- Permanently epigenetic marking - DNA methylation
PacBio SMRT sequencing
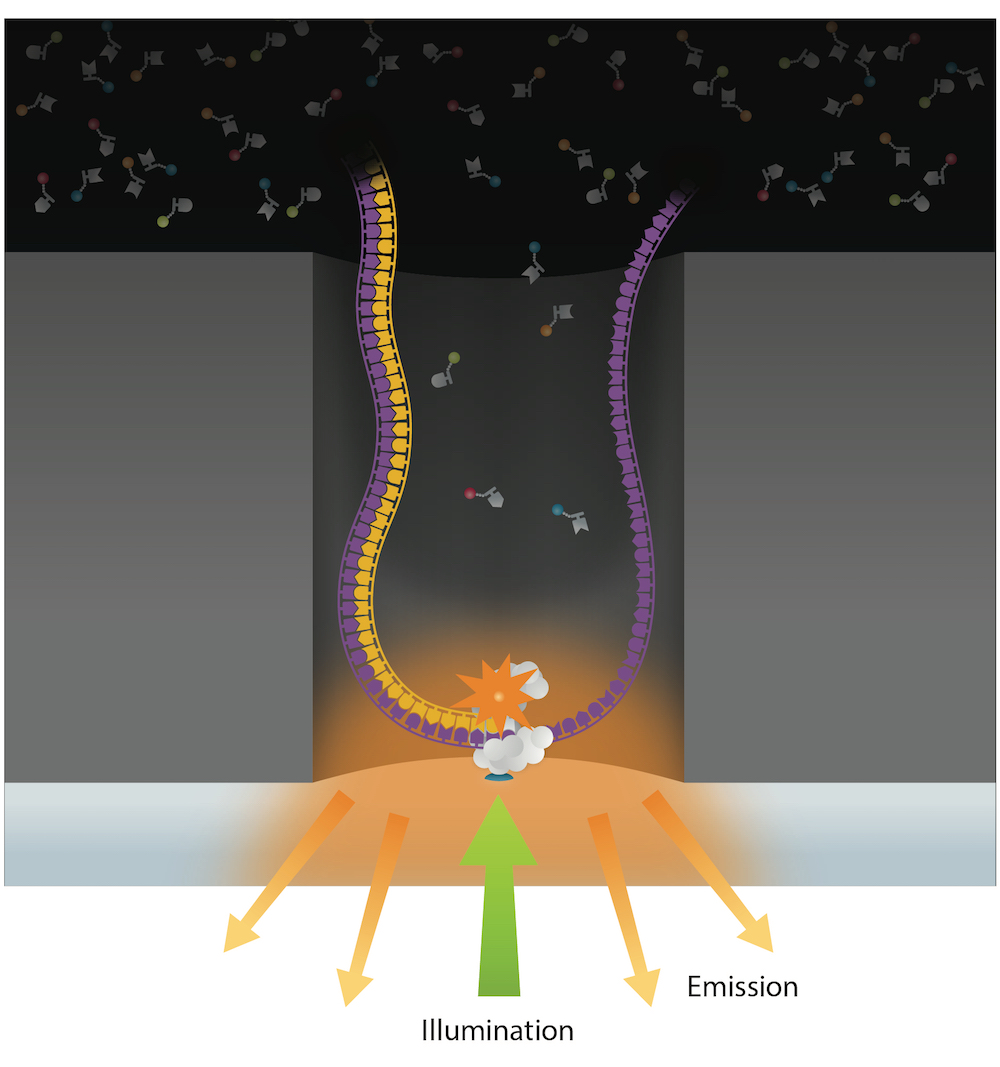
- Inter-Pulse Duration (IPD) ⇈ if n6mA
- Compared with Control or in-sillico control --> IpdRatio
- Each molecule --> analyzed independantly
- Each nucleotide --> Analyzed independantly
- Each strand --> analyzed independantly (then paired)
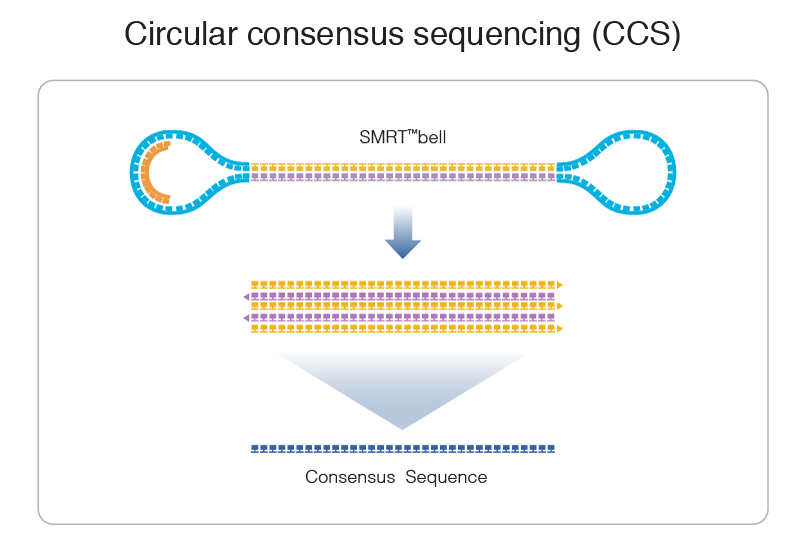
> 99% accuracy
~15% error
Strategy:
1:200
Random sampling
PacBio SMRT
- 1 out of 200 comes from the MIC
- 1/6 of MIC inserts will carry an IES
- 1/2 of IESs are just wrongly excised
- 30% of the remnants are scanRNA dependant
Only a few remaining: ~ 10 to ~100 sequences
If 100% carry a methylation pattern, this is enough
Sorting
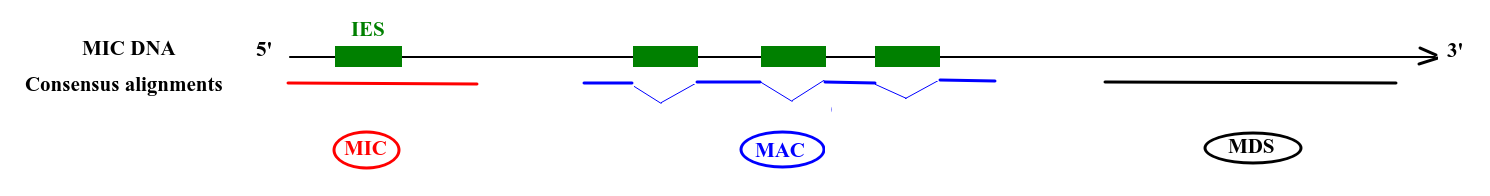
Deduced origin
MIC DNA
Alignment of consensus

MAC, MIC,
Mito...
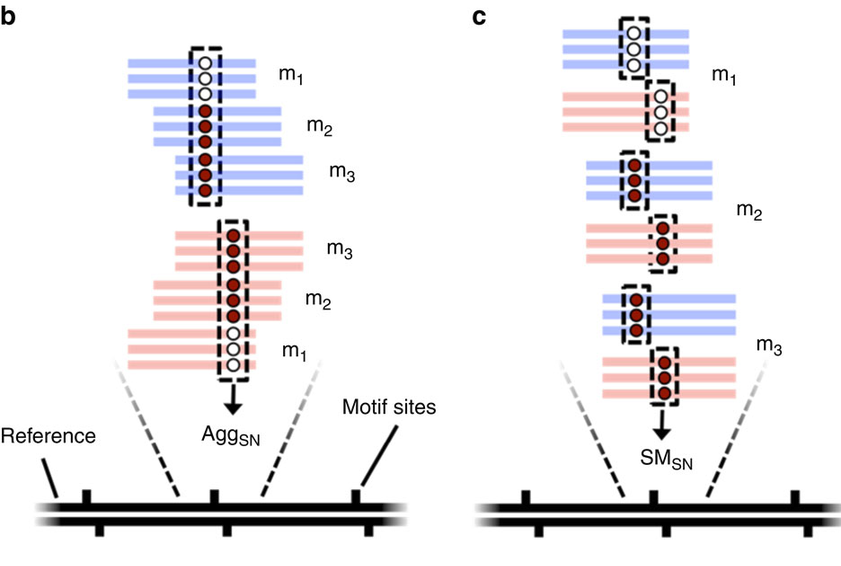
Analysis
Report n6mA
5mC, 4mC
Re-alignment
Available data at day0
Wild type:
- Vegetative Cell (HTVEG)
- During autogamy (after 2h - HT2)
-
During autogamy (after 6h - HT6)
-
Asynchronous cultures / Quite blurred state
-
Asynchronous cultures / Quite blurred state
Silencing of methylase candidates:
-
Si/MAB (identified since then - probably a histone methylase)
-
MT proteins:
- Si/MT2
- Si/MT1A-1B
-
Si/MT1A-1B-2
-
NM proteins:
- Si/NM4-9-10
- Si/NM9-10
- Si/NM4
Determining Se and Sp
-
Sensitivity, Specificity of n6mA detection ?
-
No data for our approach in the litterature
- Short inserts
- Sequel vI.0
- Can't afford a real benchmarking
-
No data for our approach in the litterature
-
Paramecium is fed with E.coli !
- ~ 100% n6mA in GATC
- EcoK1 methylation well documented
- Benchmark on E.coli ?
Found:
- Se ~ 93% = P(D+ | M)
- Sp ~ 99.9% = P(D- | NM)
Detected level VS real level
$$p= [ Se \cdot \pi + (1 - Sp) \ (1 - \pi) ] \cdot N$$
True positives
False positives
p : Number of D+
N: Number tested
$$\pi = \frac{\frac{p}{N} - 1 + Sp}{Se-1+Sp}$$
--> From imperfect detections, allows to estimate the fraction of nucleotides truly methylated
$$\pi$$ is the true proportion of n6mA
Quantitative estimation of the n6mA in the MAC
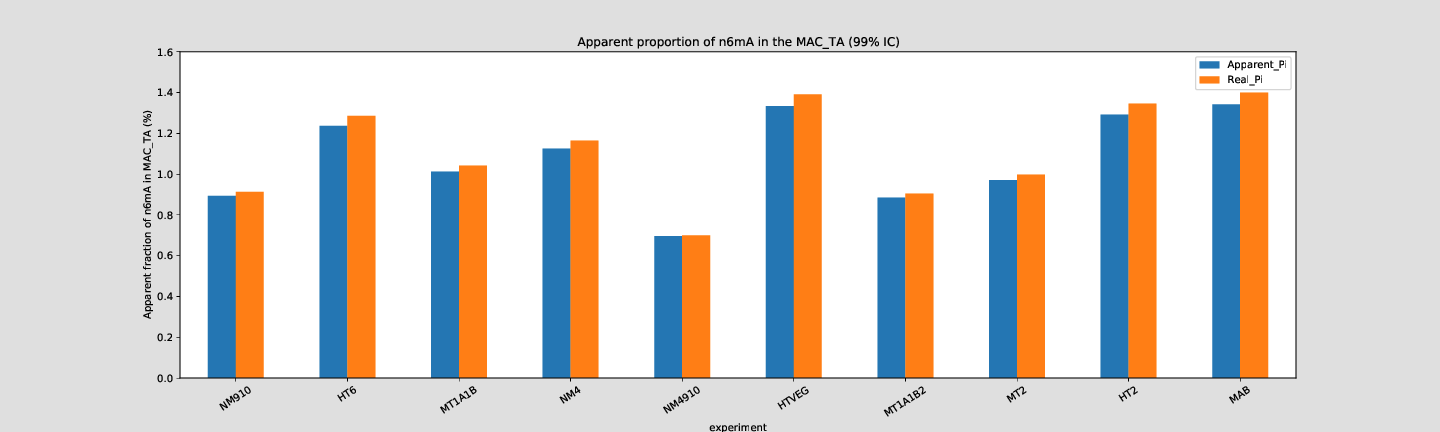
Mostly in AT sites (~90%)
"Lots" are symmetrical (~80%)
Quantitative estimation of the n6mA in the MIC
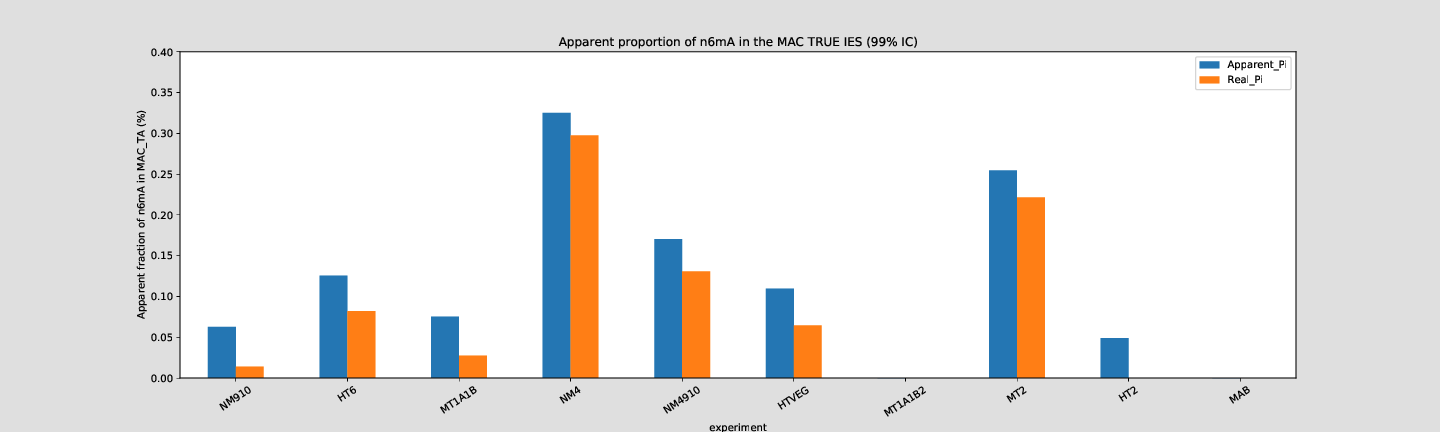
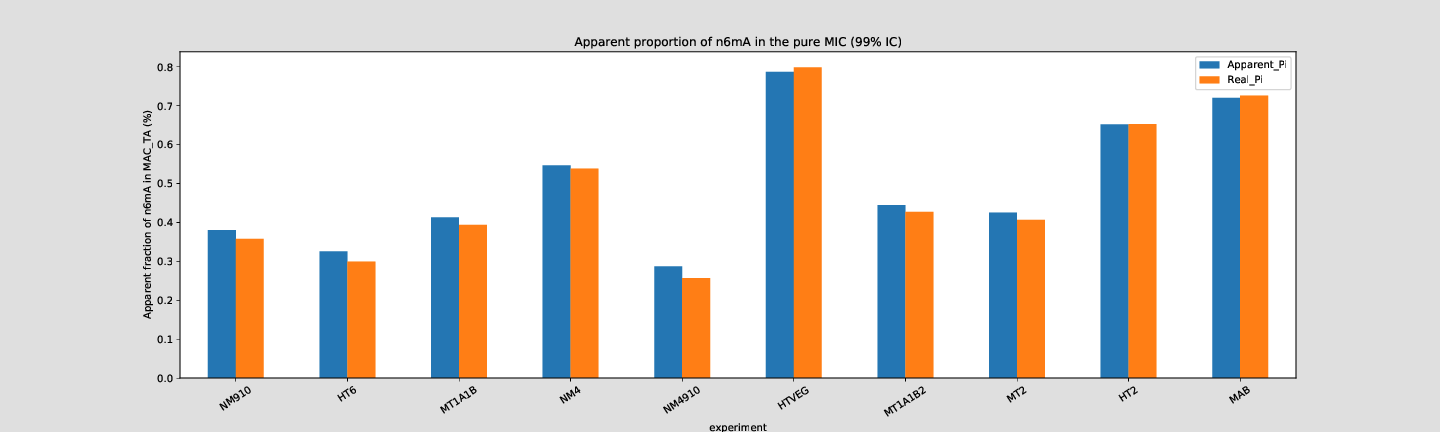
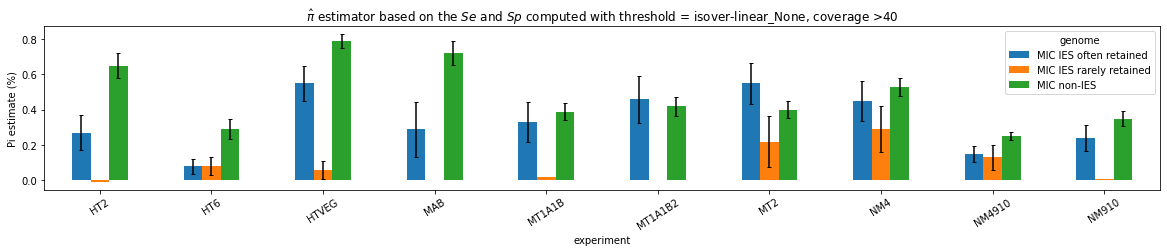
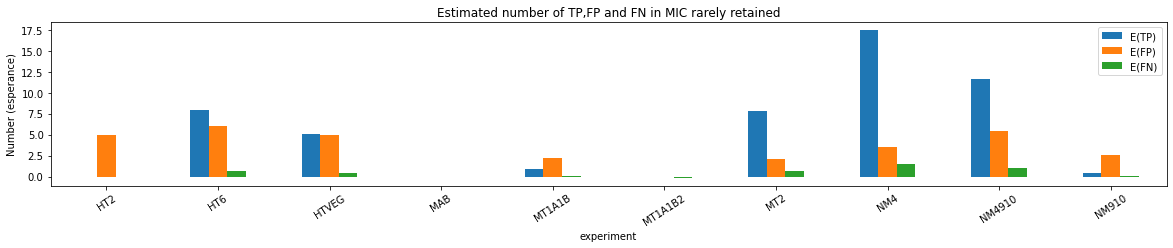
Quantitative estimation of n6mA in the MIC (details)
Distribution of the detections

HTVEG

MT2
etc...
--> Some molecules carry all the detections, in sym-A*T
Very likely to be sequences comming from the MAC
What else could we explore ?
In the MIC:
- Other type of DNA methylation
- Kinetic signal around the IES
- That's it
What else could we explore ?
In the MAC: Hemimethylation

Thanks !
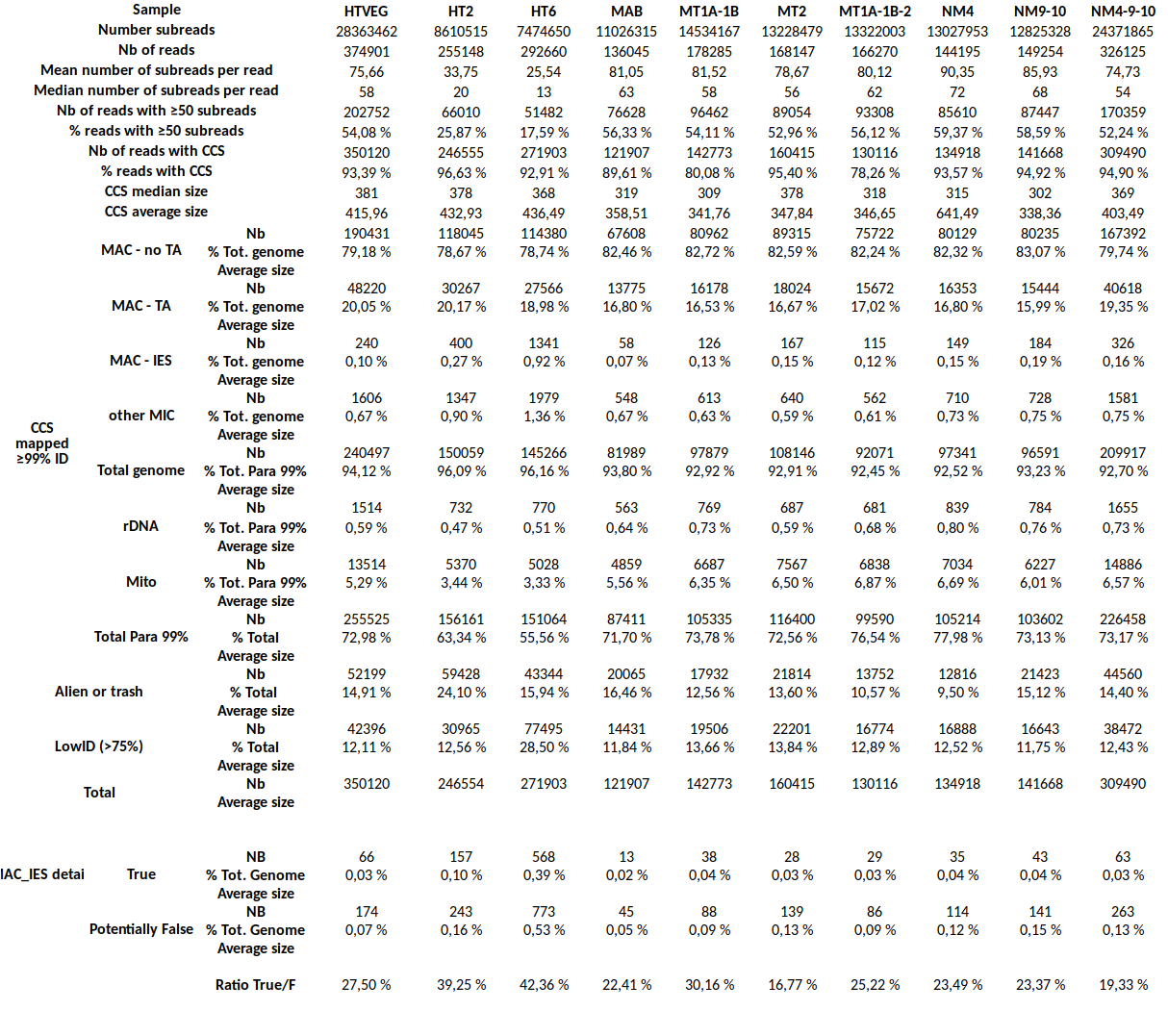
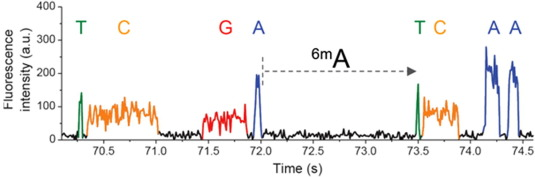
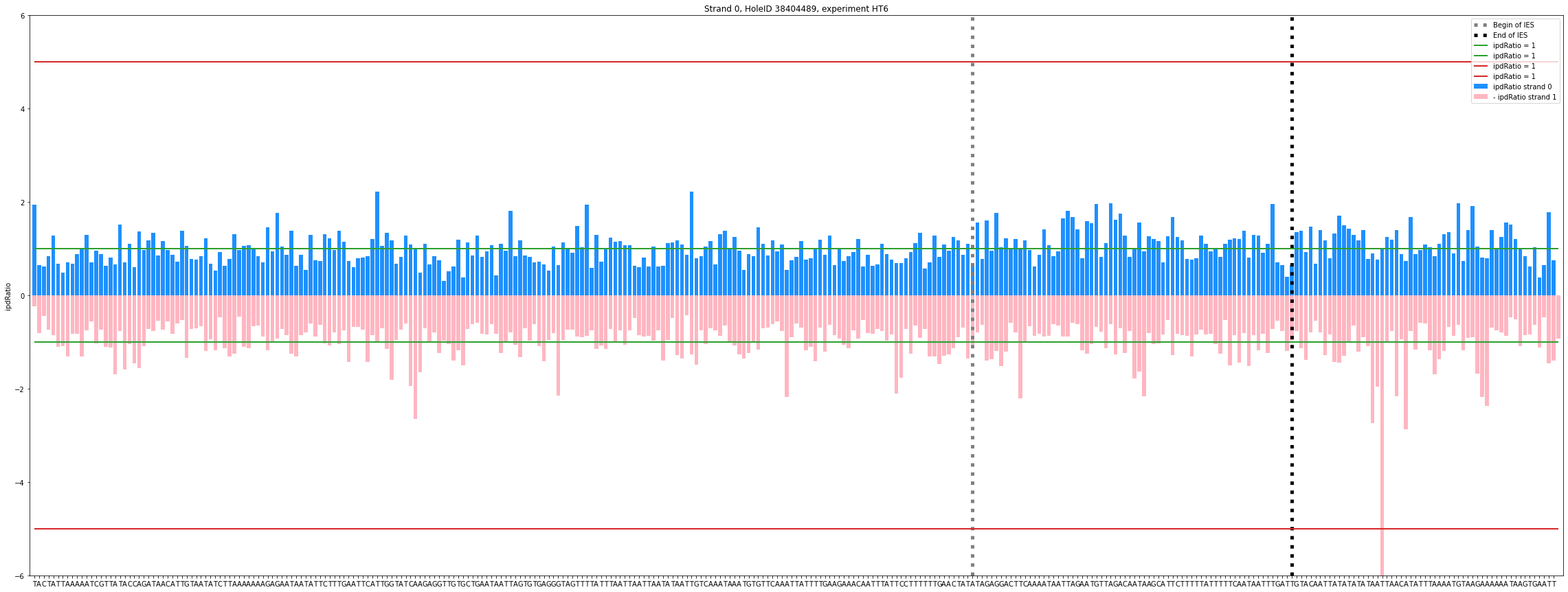
Output example
DNA n6mA
Analysis of Paramecium tetraurelia
Enfin !
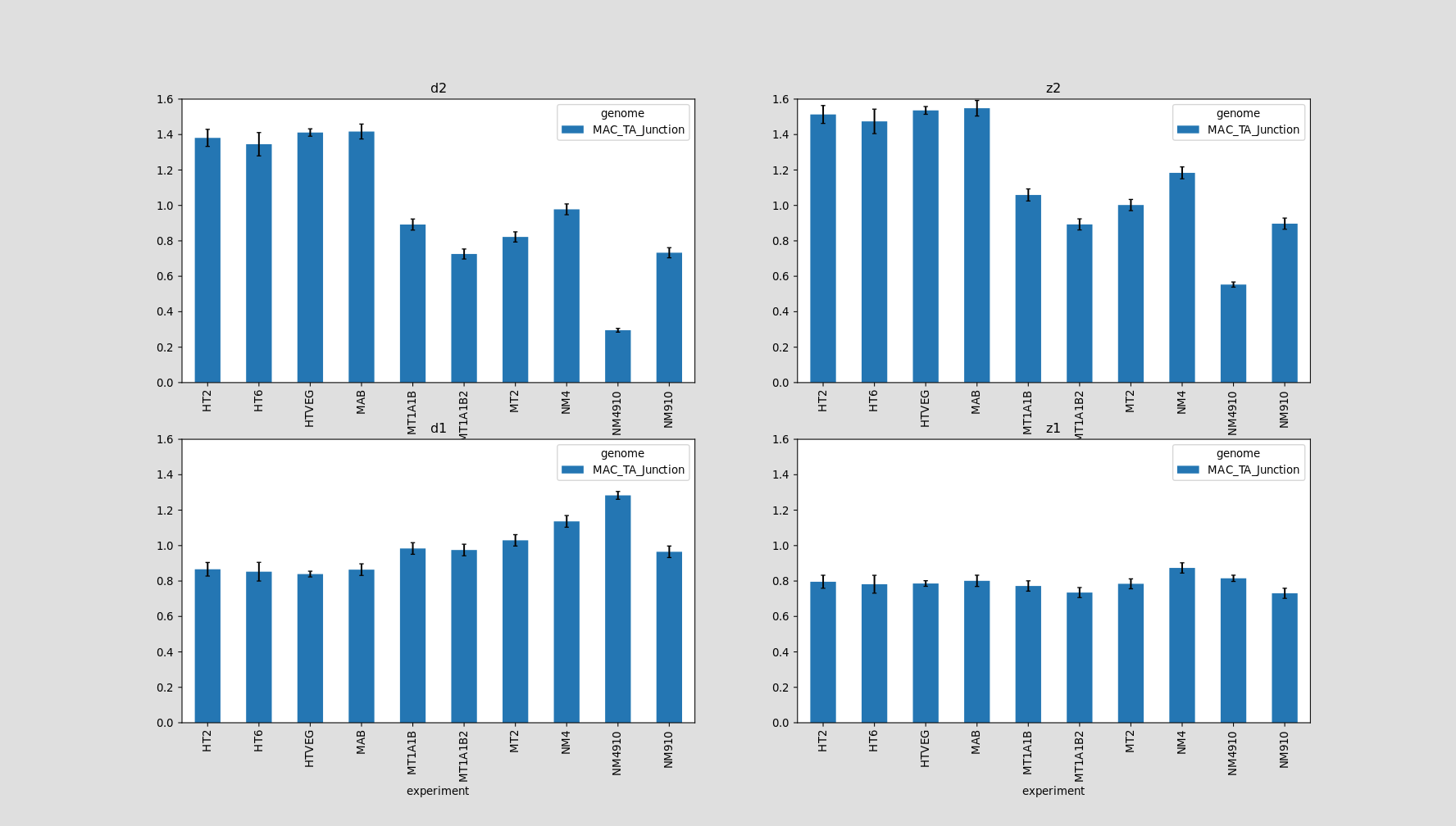
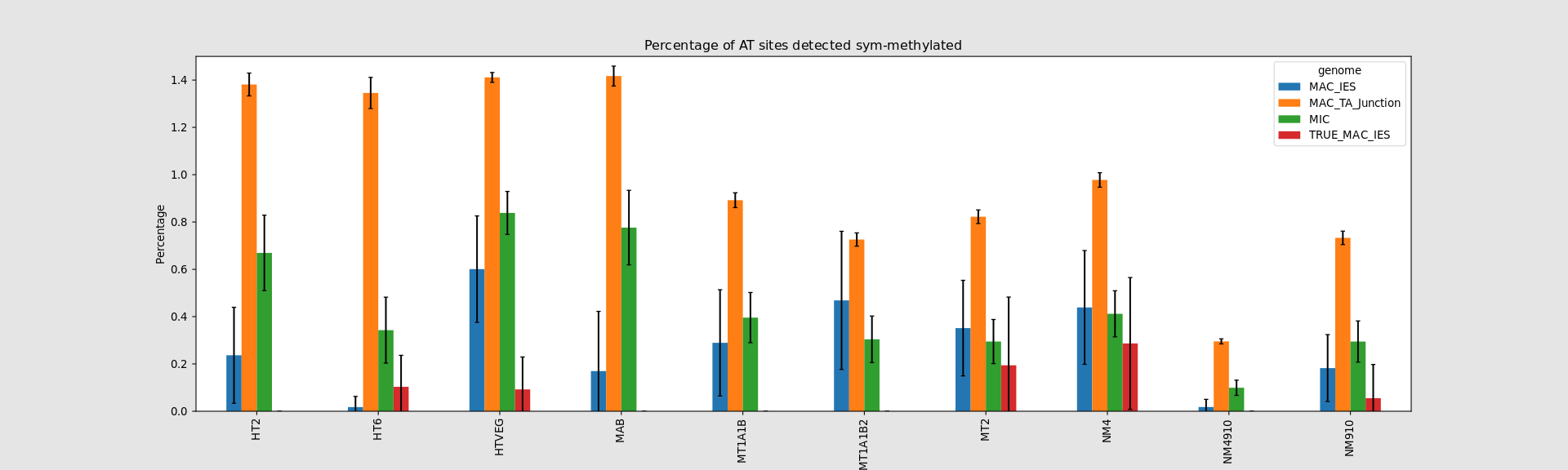
Per experiment comparaison (1)
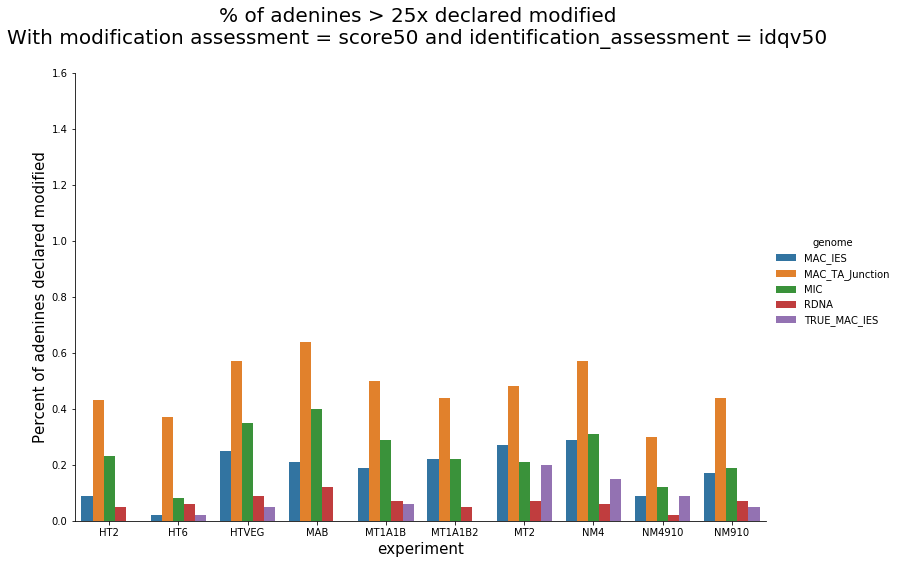
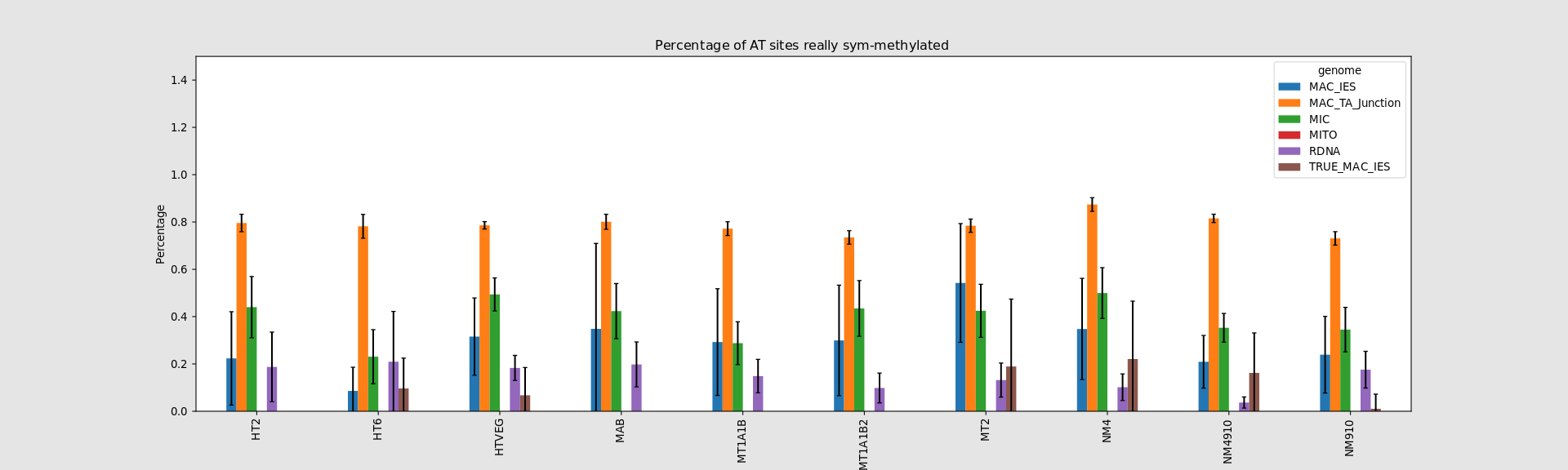
Per-experiment comparaison (2)
In the vegetative MAC
-
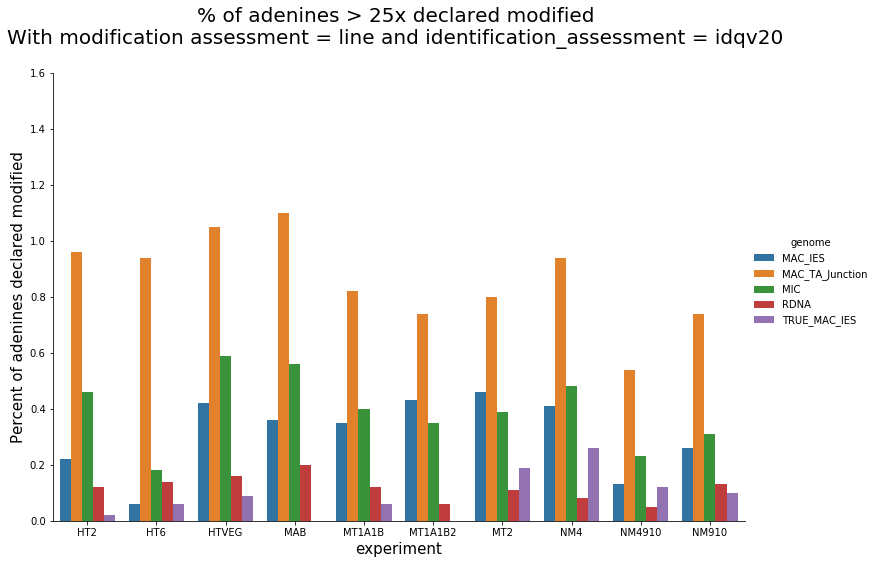
~95% of the methylation locates in AT dinucleotides in the MAC(*)
-
slightly lower in the MIC (5 to 5 points less)
-
True in any condition
-
-
75% of the methylation in an AT dinucleotide is actually symetrically modified, independantly from being in the MAC or the MIC(*)
Kept in MIC and MAC (All conditions)
(*) Linear equation / idQv20
Outside AT sites
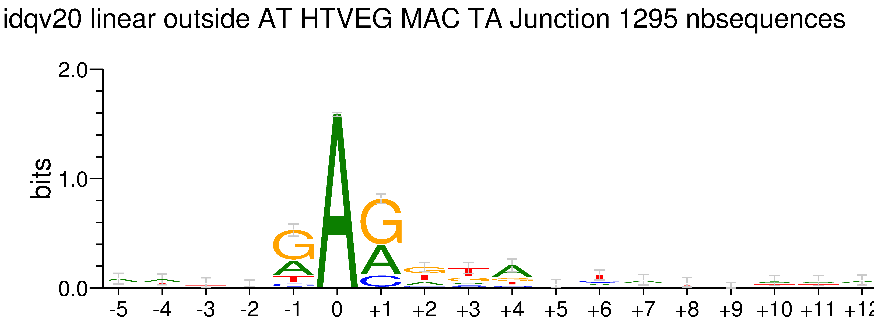
Kept in the MAC for all experimental conditions
Impossible to tell in the MIC (not enough sequences)
In the MAC
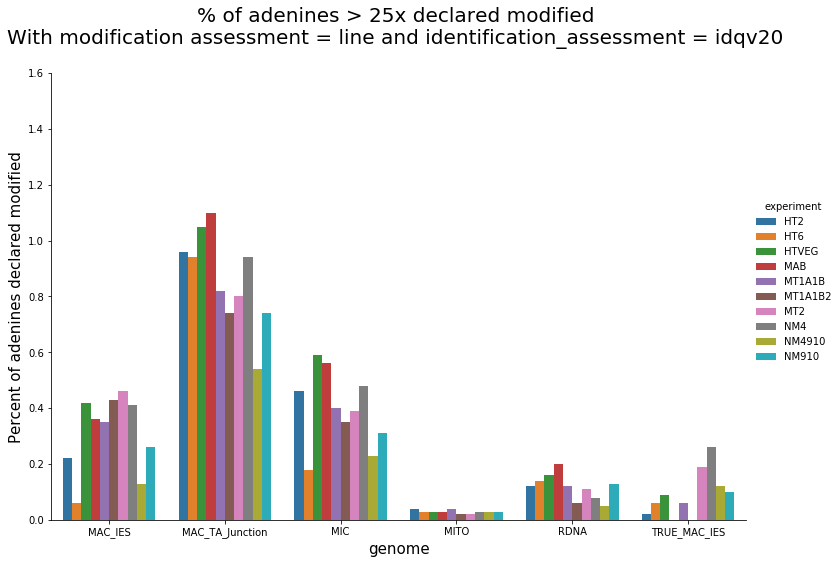
Per genome comparaison
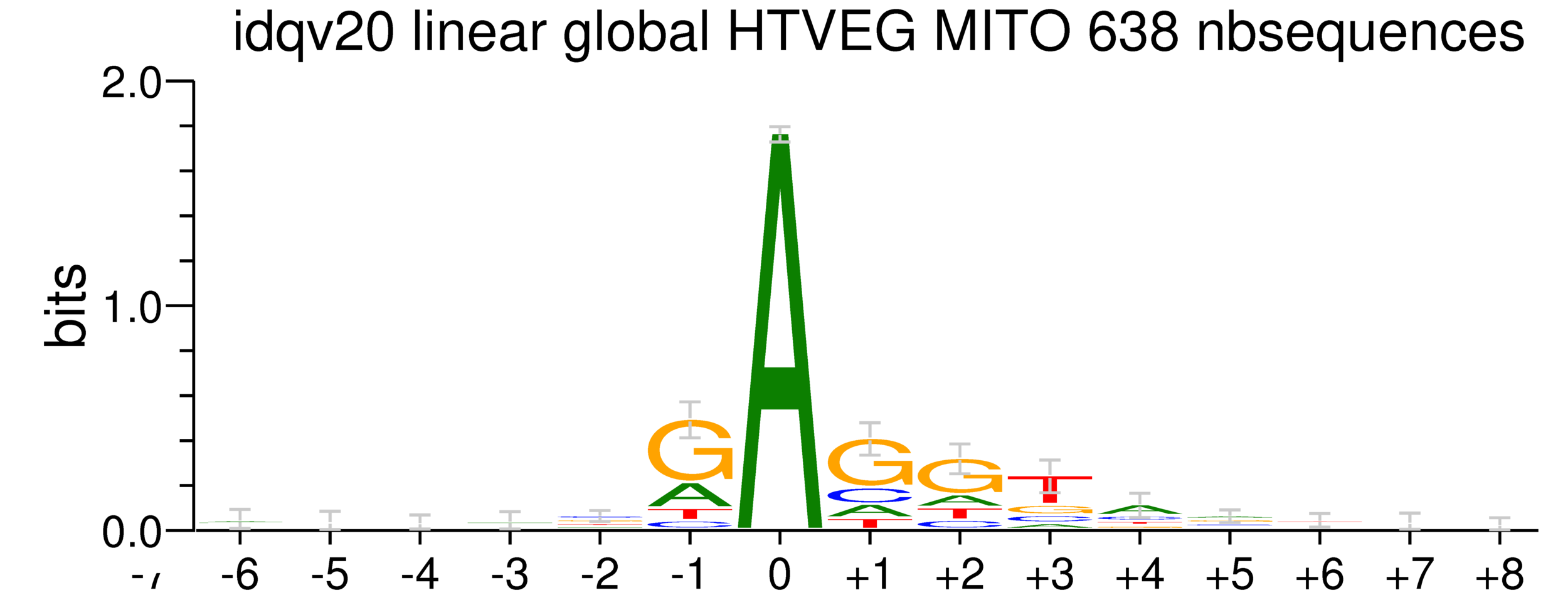
mDNA
42% GC
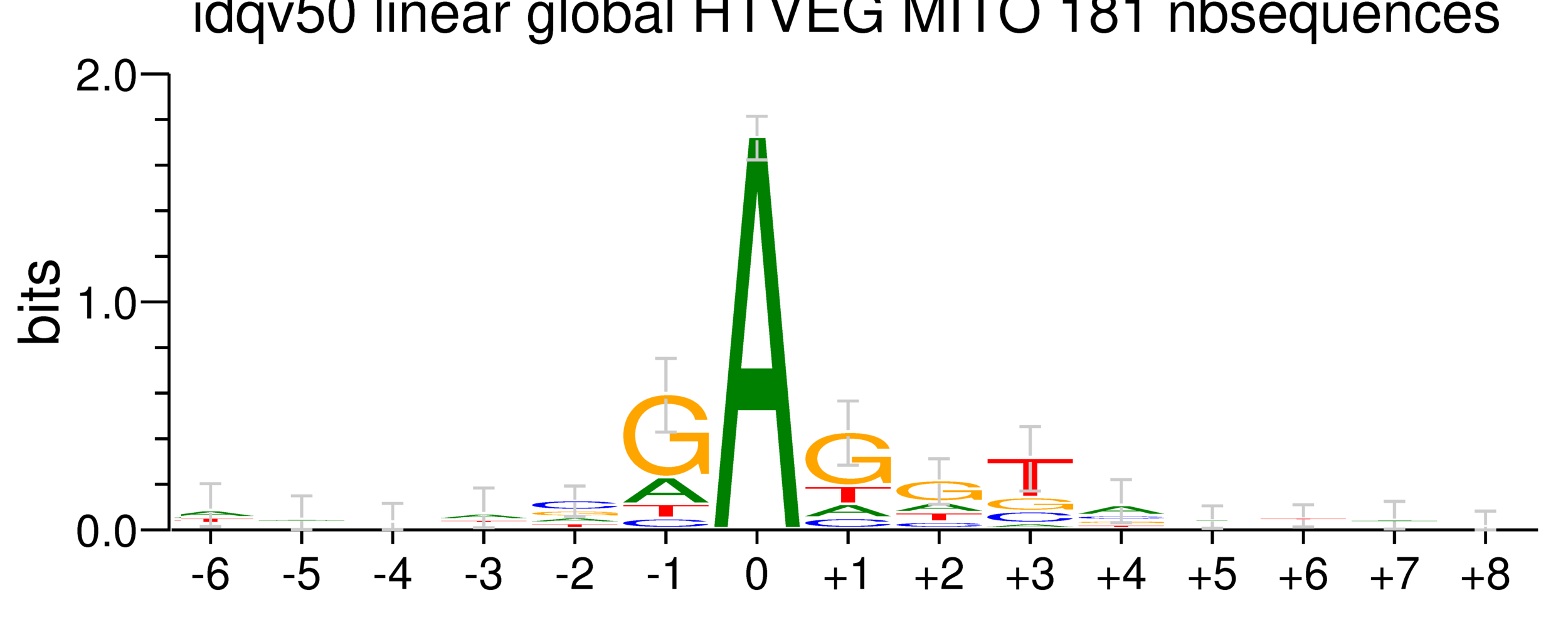
rDNA
38% GC
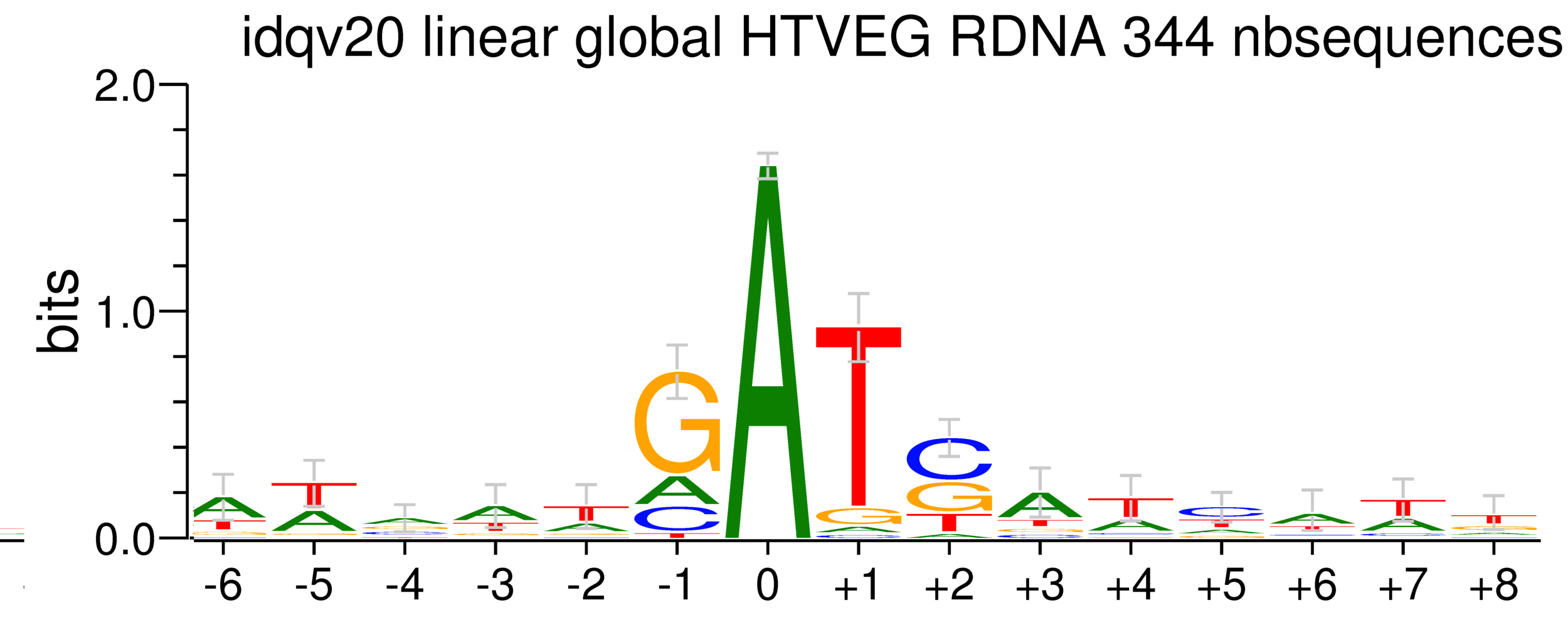
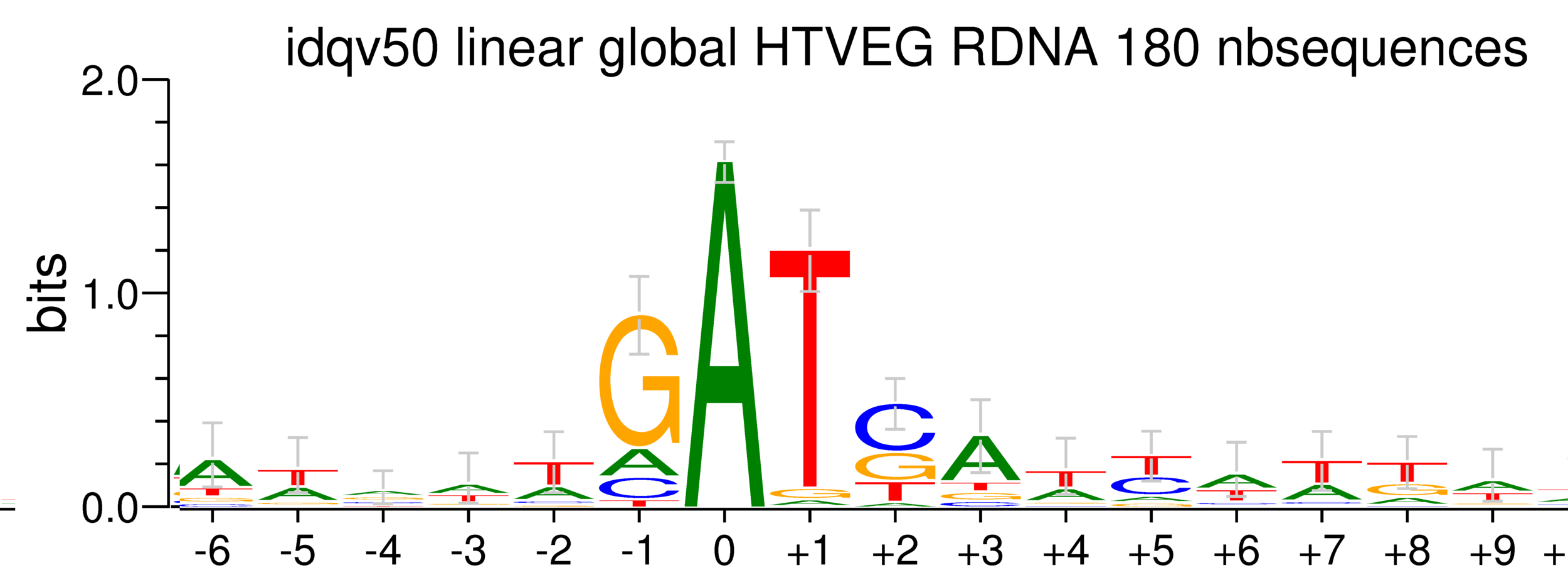
How could IES be recognized ?
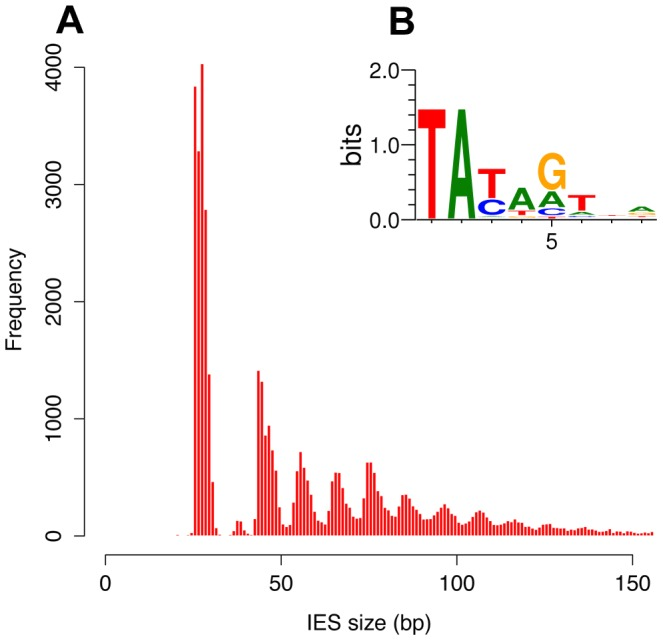
-
Weak consensus TAYAG
- Not sufficient to recognize the IES
- Degenerated TC1-Mariner TE ?
- Periodic distribution of size
~ 100% TA bounded
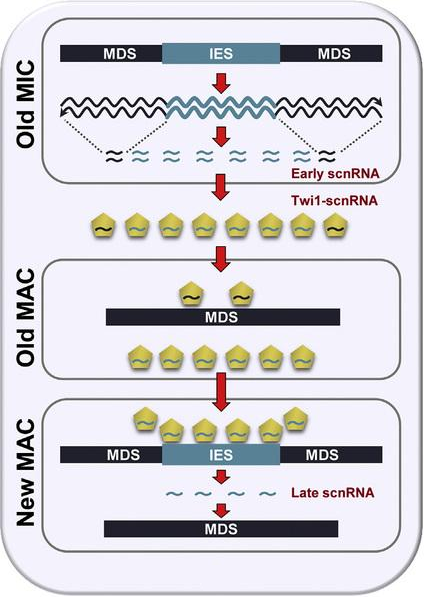
Small-RNA (~30% of IESs)
Output example