Protein-Protein Docking
with F2Dock and ZDock
Author: Kevin Song, material from Bajaj (UT) and Liwang (UC Merced)
What is a Protein?

What is the Protein Docking Problem?
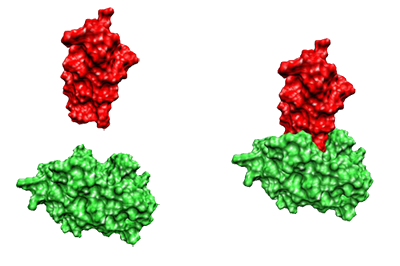
Given two proteins, find the optimal pose (positioning) that maximizes bonding interactions
Factors to Consider
Properties of a good dock
- Surfaces close together
- Opposite charged atoms close together
- Hydrophobic surfaces hidden
Properties of a bad dock
- Proteins tunneling into each other
- Same charges close together
Goal: Given two protein structures, optimize docking
If proteins were rigid bodies, this problem would be (relatively) easy! Unfortunately, life is complicated...
Meet Hemoglobin!
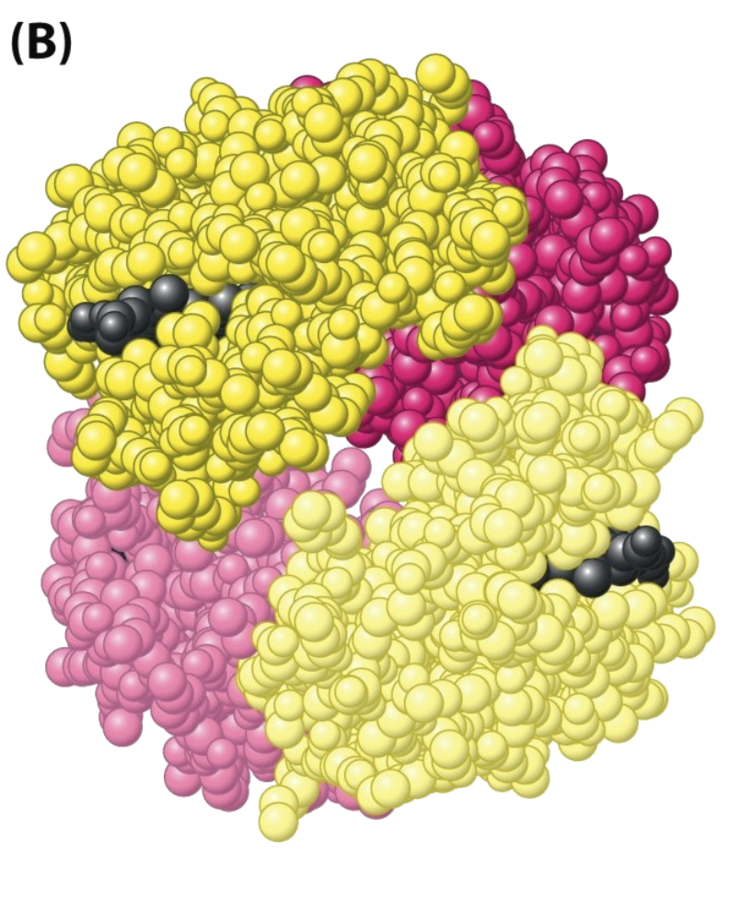
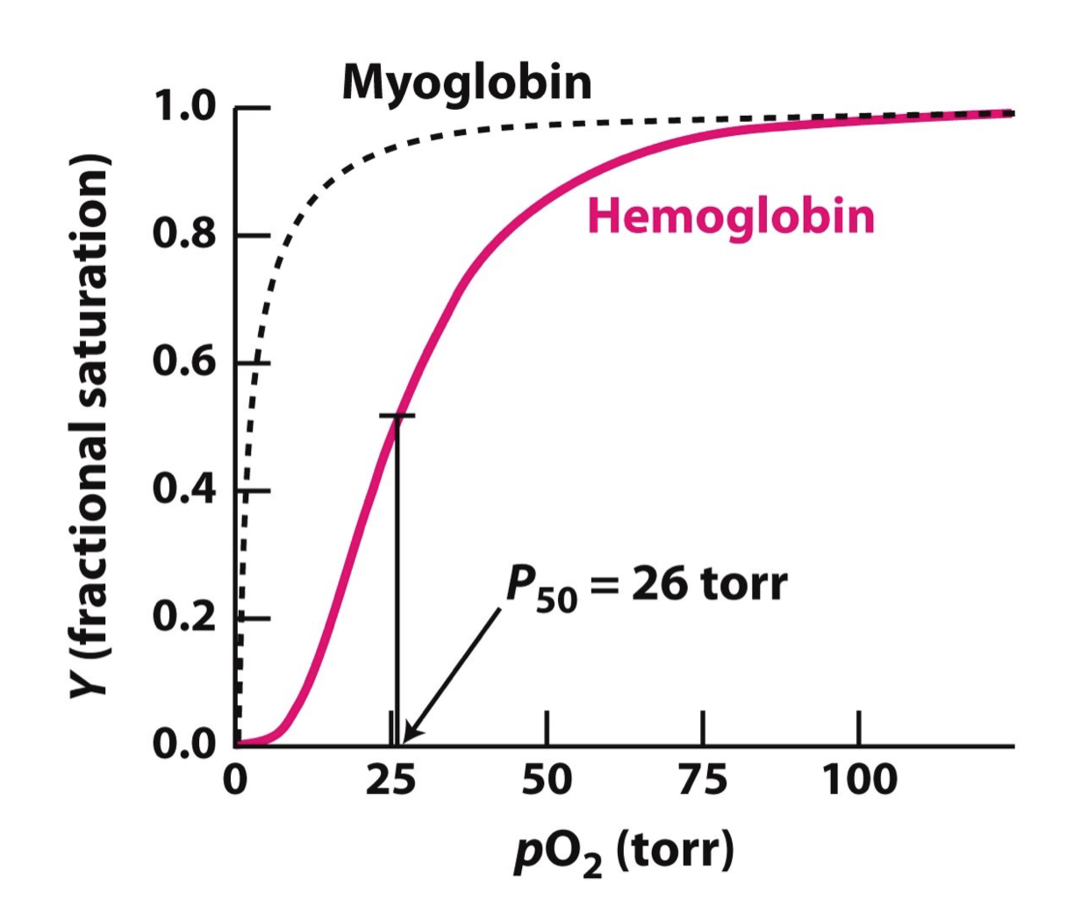
Images from Berg,Tymoczko,Stryer, 8th ed.
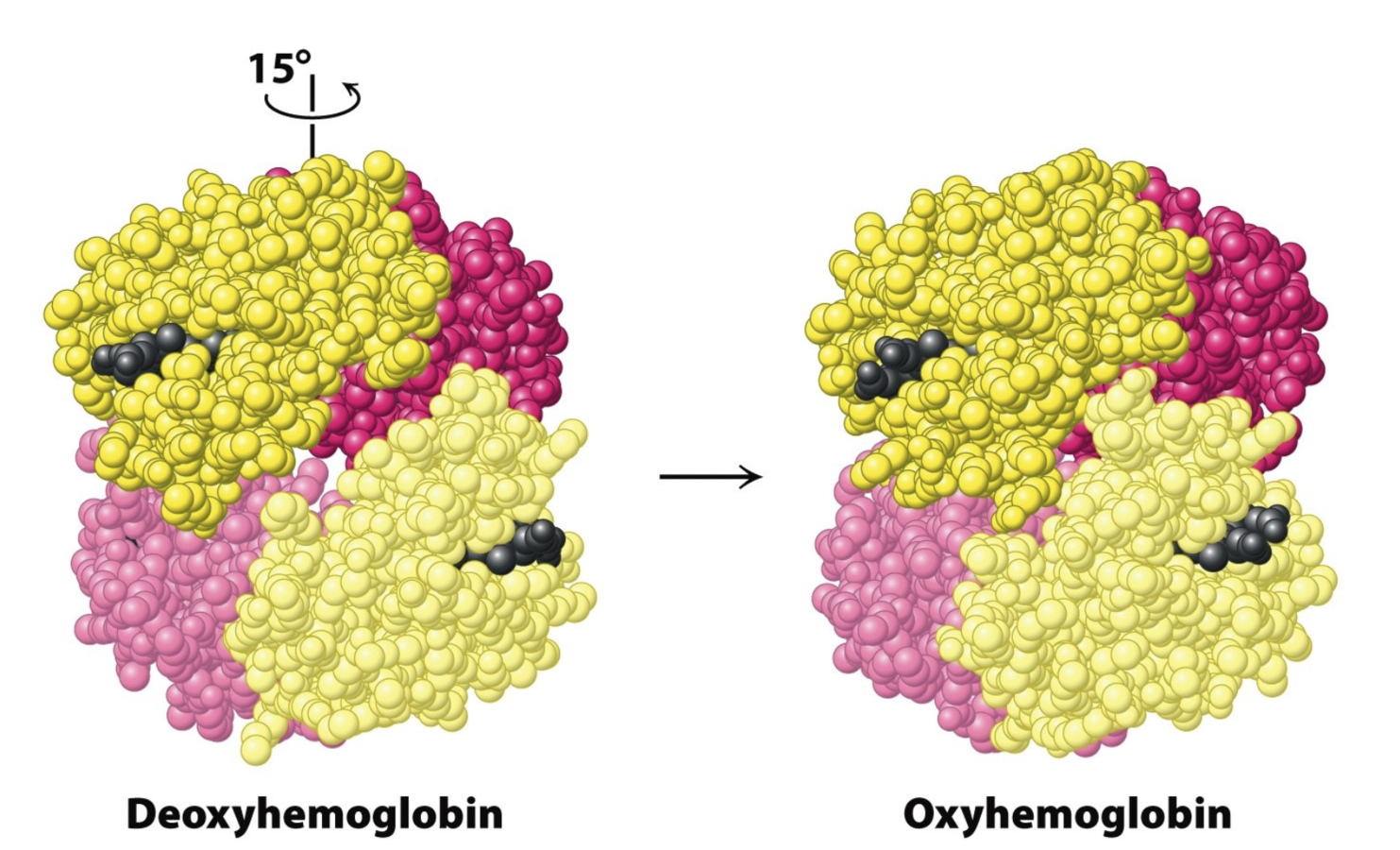
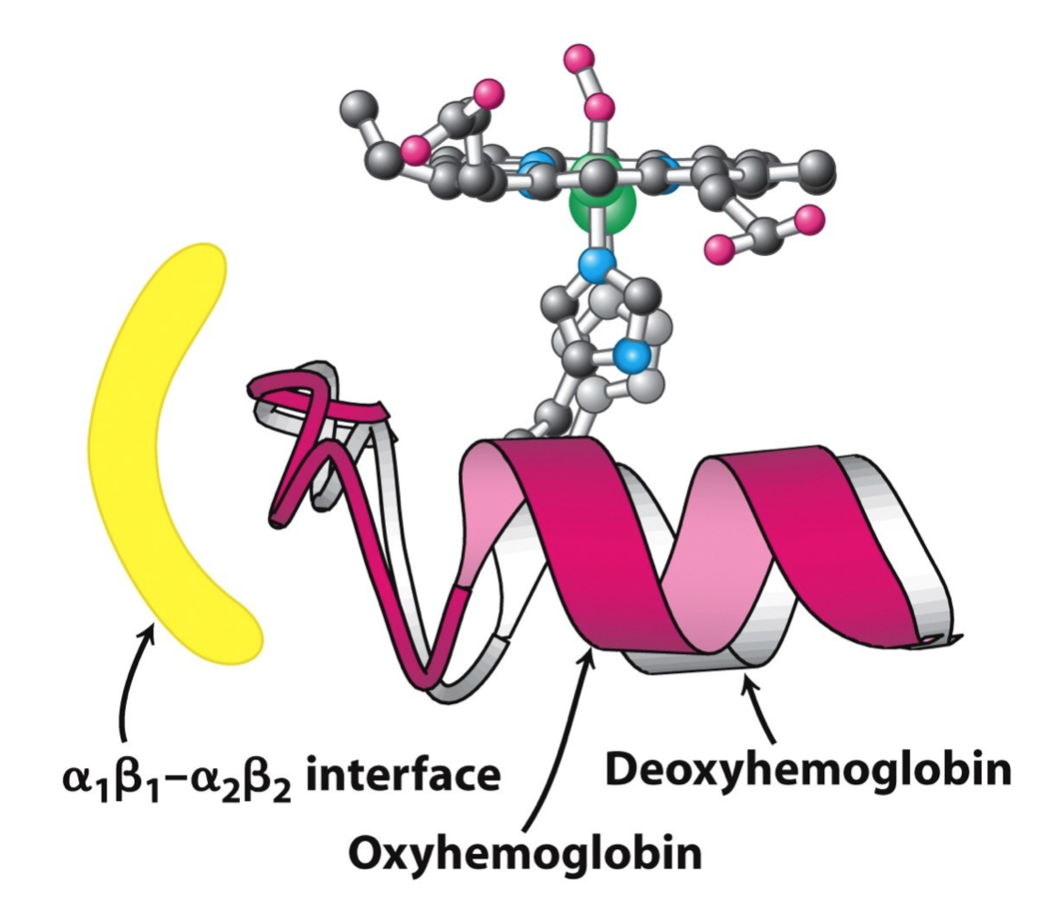
Oxygen binding in deoxyhemoglobin stimulates conformational change. Oxyhemoglobin has better affinity to oxygen.
Images from Berg,Tymoczko,Stryer, 8th ed.
Other Factors at Play
- 2,3-bisphosphoglycerate in
quaternary binding pocket - Amino acids in α2γ2 subunit affect
oxygen affinity - Protonation of residues (Bohr Effect)
- Additional effects (not well understood)
Protein Binding is complicated!
Simulation Techniques
Molecular Dynamics
- Use physics (force fields and energy) to calculate molecular motions
- Can incorporate elements of Monte Carlo simulations (randomness)
- Is the gold standard for most protein problems
Literally Everything Else
Soft Optimization Methods
Principal Idea: We can't compute all conformational changes that occur upon binding.
Instead, find some objective functions that tolerate a little bit of mismatch, and hope that this allows us to find docking sites.
Problem Statement
Given two molecules and , and an affinity function , find all rotations and translations such that:
where is a user-defined threshold and and are weights
Affinity Functions
Define an Affinity Function associated with a molecule to be a function from
For any two proteins A and B, the docking score) can be evaluated by the following integral:
Example Molecules
A
B


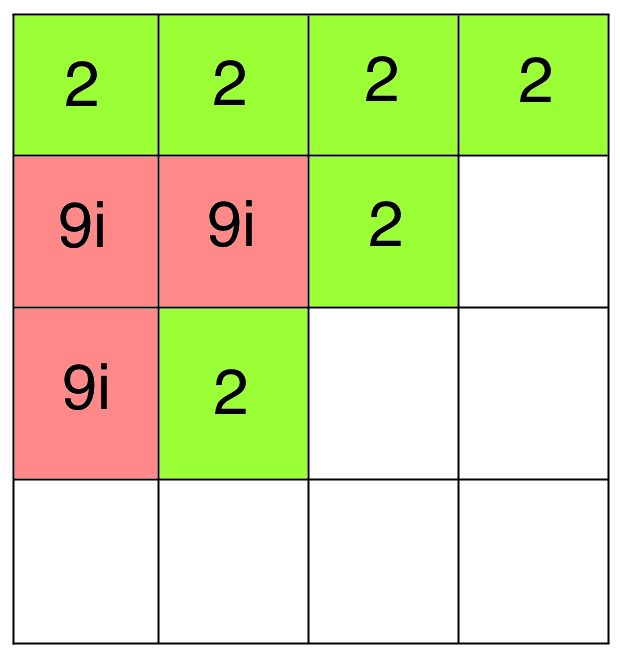
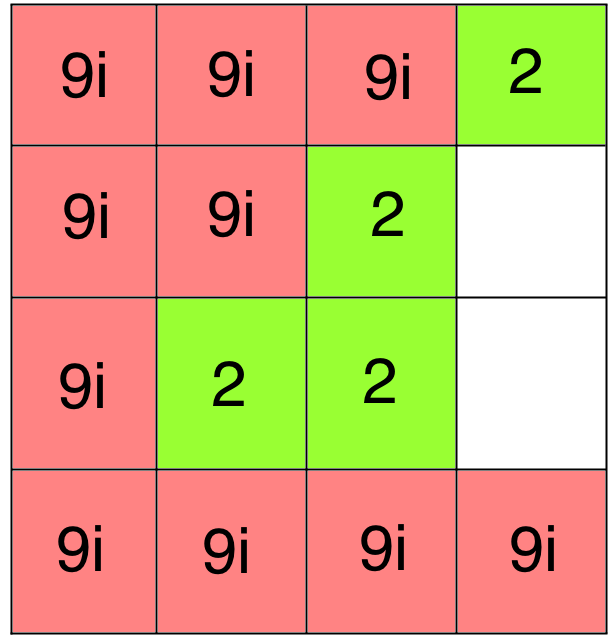
Bad Docking!
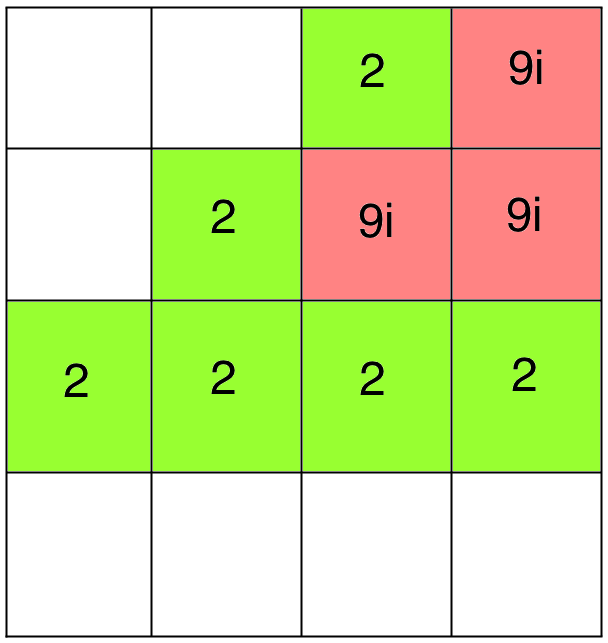
Good Docking!
Problem Statement
Given two molecules and , and an affinity function , find all rotations and translations such that:
where is a user-defined threshold and and are weights
Real Affinity Functions
Shape Complementarity
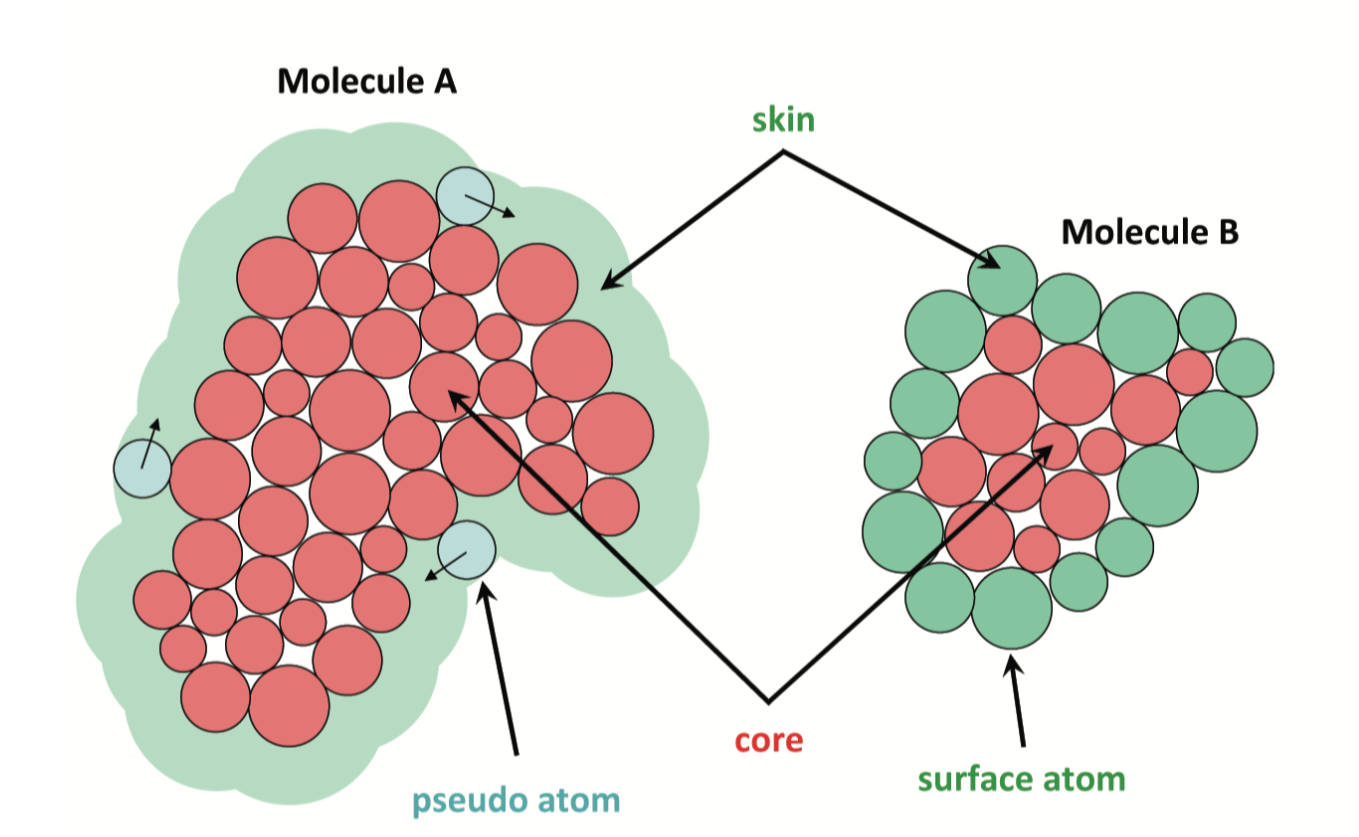
For Receptor: Positive weights in grown-skin layer, imaginary weights in core.
For Ligand: Positive weights in surface layer, imaginary weights in core.
Real Affinity Functions
Electrostatics
Coulombic Potential Energy
So how do we actually find the best configurations?
Naïve Method
For every possible translation and rotation, evaluate the integral (or summation) of affinity functions.
In a 3D grid with N intervals, it takes time to evaluate the summation once.
There are possible translations.
With rotations, this is a search time of
Look at 1D Case
Want to find the offset such that the overlap integral is maximized:
This integral is a cross-correlation!
Look at 1D Case
The Fourier Convolution theorem allows us to express the convolution as a product of its Fourier Transforms!
Naïve 1D Convolution
1D Fourier Convolution
Use FFT to solve 3D
We can convolve the affinity functions along the translations and perform an inverse peak search to rapidly identify translations for which the score is above the threshold!
Convolution with FFT in 3D
3D FFT
Peak Search
Total Runtime
Much better!
ZDock
ZDock is an older protein-docking software suite that was developed at BU
Grid-based affinity functions:
- Shape Complementarity (GSC)
- Desolvation Energy (ACE)
- Electrostatics (CHARMM19)
Later versions replaced GSC with a better shape-complementarity function (PSC) and added faster FFT routines.
Pierce BG, et. al. (2014) ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers.
ZDock PSC
Pierce BG, et. al. (2014) ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers.
An affinity function based off of number of nearby atoms on the other protein.
In a naive setting, much slower to compute than traditional grown-skin shape-complementarity functions.
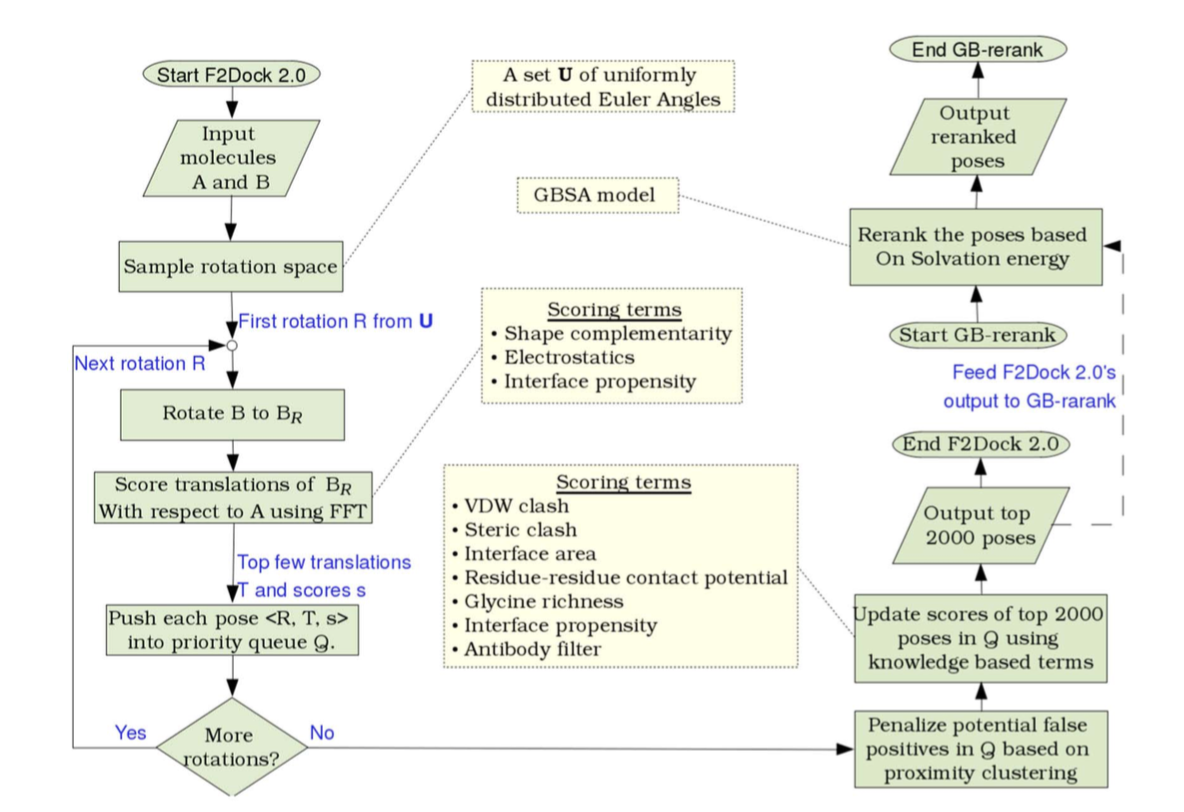
F2Dock
A relatively recent soft docking suite developed by CVC @ UT.
Saves time and memory by using NFFT (nonequispaced FFT) and exploiting sparsity in the inputs and outputs.
C. Bajaj, R. Chowdhury, and V. Siddavanahalli F2Dock: Fast Fourier Protein-Protein Docking
Only uses SC and Electrostatic AF!
F2Dock
After initial ranking, cluster poses and rerank with Generalized Born solvation energies
C. Bajaj, R. Chowdhury, and V. Siddavanahalli F2Dock: Fast Fourier Protein-Protein Docking