BIOSC 1540: L03A (Genome assembly)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Jan 21, 2025
Lecture 03A
Genome assembly
Foundations
Assignments
Quizzes
CBytes
ATP until the next reward: 1,903
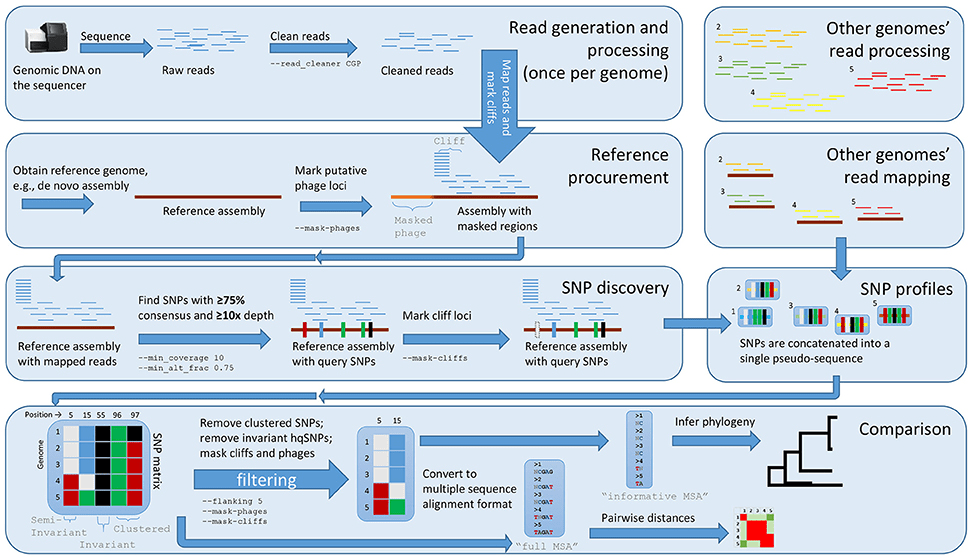
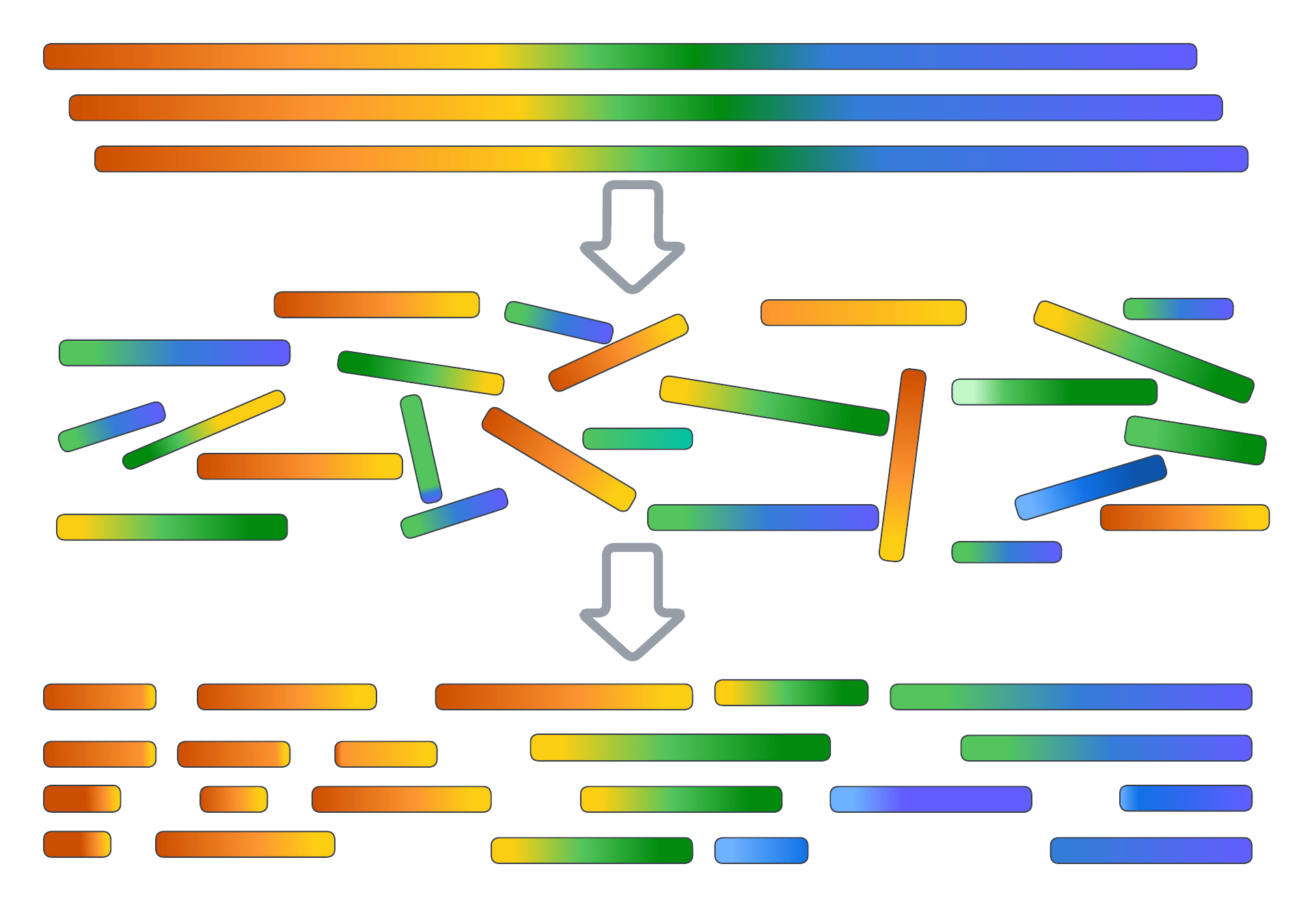
Modern sequencing technologies generate millions to billions of short reads (i.e., DNA fragments) from a DNA Sample
Reads are typically 100–300 base pairs long for short-read technologies and up to tens of kilobases for long-read technologies
DNA sample of unkown sequence
Reads
Sequencing
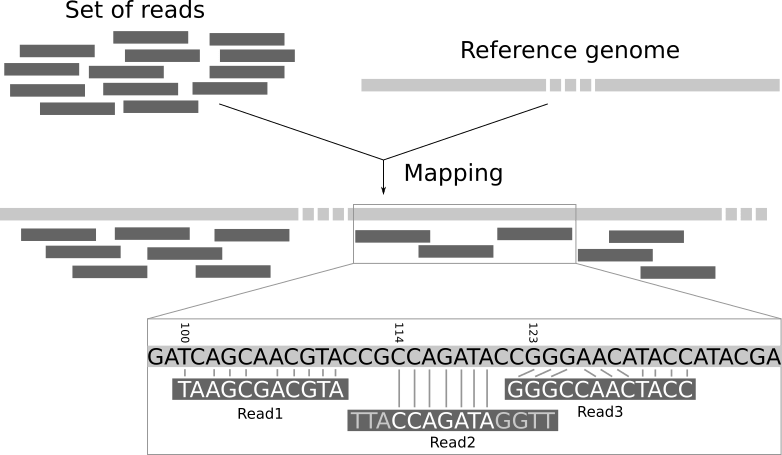
Reads overlap where they represent the same genomic region
Assembled DNA sequence (i.e., contig)
Reads
Assembly
Overlap information is used to merge reads into contiguous DNA sequences called contigs
Assembled genomes are essential for identifying genes and understanding regulatory elements
Provides a foundation for downstream analyses, including functional and structural genomics
Reference-based
Sequencing reads are matched to a reference genome to determine their correct positions
Alignment relies on identifying overlaps and shared sequences between the reads and the reference
RefSeq provides high-quality reference genomes, transcriptomes, and proteins
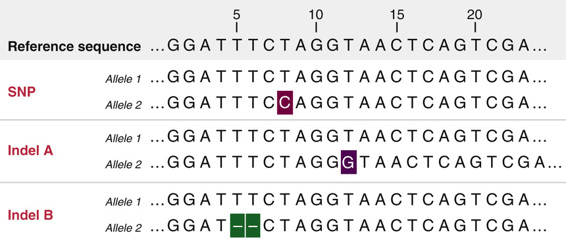
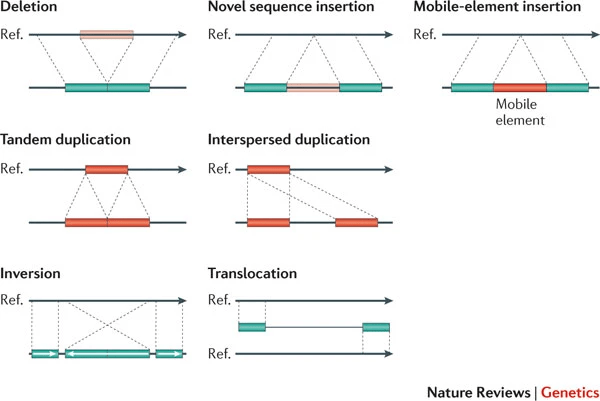
Variations occur when a read differs from the reference genome
Single-nucleotide polymorphisms (SNPs) are single base changes between the read and the reference
Indels are small insertions or deletions that alter the alignment pattern
Example: GRCh38.p14 for Humans
Reduces time and cost for studies focused on variant detection or evolutionary comparisons.
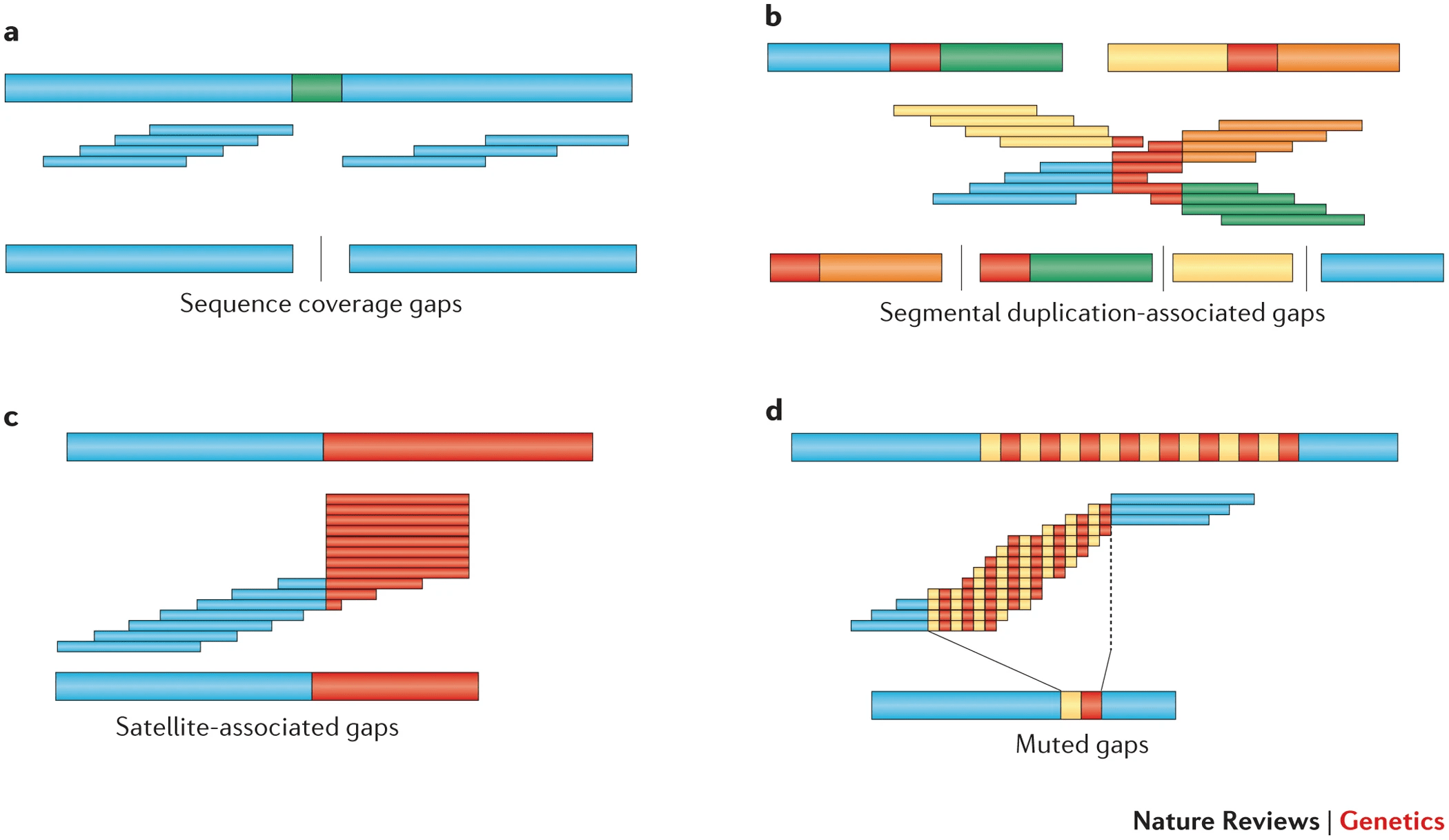
Reads corresponding to regions missing in the reference cannot be mapped, leaving unassembled gaps.
Reads
Missing gap
in reference
Inaccurate assembly
These gaps can affect downstream analysis, especially for novel genes or functional elements.
Gaps can occur due to incomplete reference sequences or highly divergent regions in the sample genome.
Variations like insertions, deletions, inversions, or translocations may not align correctly to the reference.
Failure to account for structural variations can skew results and mask important genomic differences.
Assemblers may interpret these variations as mismatches or sequencing errors.
De novo
It does not rely on pre-existing data, allowing for unbiased genome reconstruction. Essential for novel organisms or those with no reference genome.
Instead of mapping to a reference, reads are assembled by finding overlaps between reads and merging them
Unbiased assembly enables the discovery of unique and divergent sequences.
Resolves structural variations that reference-based methods might miss.
Ideal for exploring non-model organisms and highly variable regions.
High computational requirements due to complex algorithms
Most methods use graph-based methods (more on this in the next lecture).
Struggles with repeats, sequencing errors, and low-coverage regions (more on this later).
Researchers are analyzing the genome of a newly discovered bacterial strain suspected to carry antibiotic-resistance genes. They have access to a draft reference genome from a closely related strain, but it is incomplete and poorly annotated. Their main goal is identifying novel resistance genes while ensuring assembly accuracy and minimizing computational costs. Which approach would you recommend for assembling the genome, and why?
A. Use reference-based assembly to ensure computational efficiency and focus on conserved regions.
B. Use de novo assembly to avoid reference bias and discover novel resistance genes.
C. Use hybrid assembly, starting with reference-based assembly and refining with de novo assembly for poorly aligned regions.
D. BLAST reads that fail to align to the reference genome but avoid de novo assembly to reduce computational cost.
Biological factors: Repetitive sequences, structural variations, and genome size.
These challenges complicate the process of accurately reconstructing a genome.
Technical issues: Sequencing errors, low coverage, and short read lengths.
Overcoming these challenges requires balancing biological and technical factor
Advances in sequencing technology (e.g., long-read sequencing)
Careful experimental design (e.g., choosing read length and depth)
Repetitive DNA (i.e., repeats)
Repeats are sequences of DNA that occur multiple times in the genome
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT
Note: Repeats are especially abundant in eukaryotic genomes, comprising up to 50% of human DNA.
Common types of repeats
Tandem repeats: Consecutive copies of the same sequence.
Interspersed repeats: Similar sequences scattered throughout the genome
AGCTGATC
TTAGCCGA
CGAT CGAT
CGAT CGAT
How will the assembler know the difference between these two options? Maybe it has high coverage instead of more repeats?
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
Reads
Option 1
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT
Option 2
Reads from repeats may align to multiple locations, making it unclear where they belong.
Which repeat did a read come from? Who knows ...
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT
Fragmentation: Assemblers may break contigs at repetitive regions, resulting in gaps.
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
versus
Collapsing repeats: Similar repeats may be merged into a single copy, leading to incorrect assemblies.
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT
Short reads: Often shorter than repeat regions, making it difficult to span and resolve repeats.
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
Long reads: Can span entire repetitive regions, reducing ambiguity and improving assembly accuracy.
Paired-end reads are sequenced from both ends of a DNA fragment, with a known distance between the reads
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
If I have two reads at the ends of a repeat, and I know the distance between the reads, I know the length of repeat
Forward read
Reverse read
Known read gap
(This is why having paired-end reads that do not overlap is helpful.)
Sequence errors
Sequencing errors interfere with overlaps by creating mismatches between reads
Assemblers must distinguish true overlaps from errors, which dramatically increases computational complexity.
Redundant data (high coverage) helps correct errors by identifying the most likely base (i.e., consensus)
TACGATCGGATTACGCGTAGGCTAGCTTACGGACTCGATGTACGATCGGATTACGCGTAGG
Real sequencing errors
can be fixed in high-coverage areas
Real SNPs
can be confidently detected when all reads have the same base
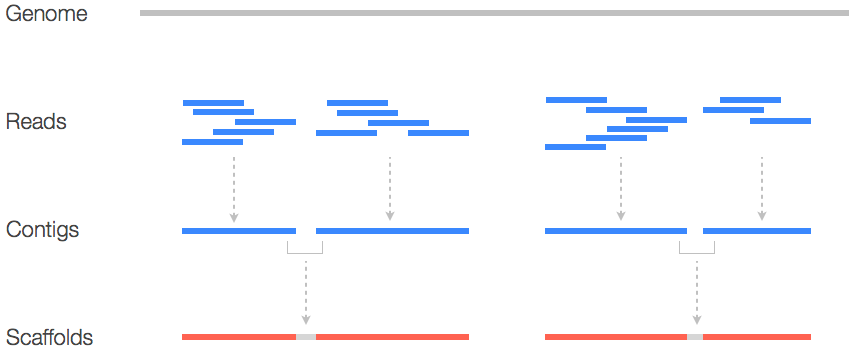
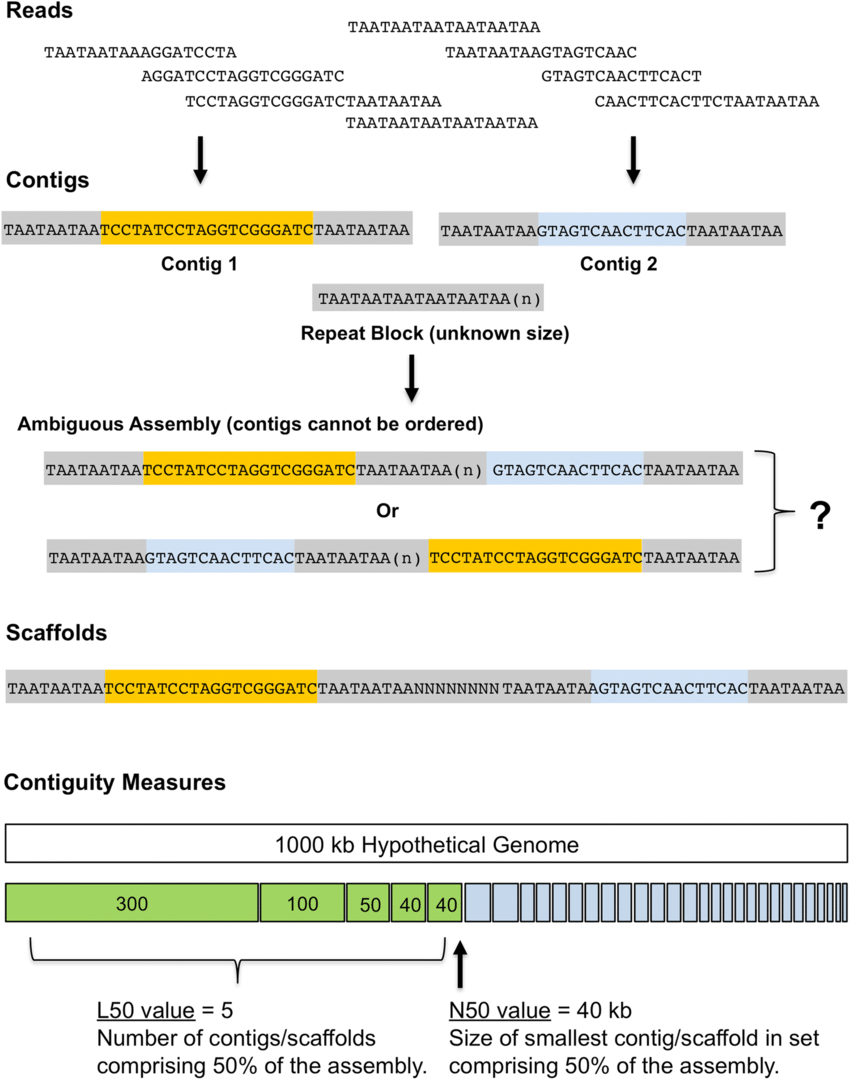
Contigs are the first level of assembly, where reads are merged based on overlaps. In other words, they represent reconstructed DNA without gaps (i.e., continuous).
What do contigs indicate?
What is a contig FASTA file?
>NODE_1_length_251580_cov_96.965763
GCCTTTTTCATATTCTTGAAACATATATAGCAGTACATCTATGTCTACTTTAGGTTTTAT
TGACATAAATAAAGCTCCCTTCAAAGTTTTCATTTTTTCAATGTCTACTTTGAAGGGAGC
ATTTCACTGAACTTTGTTCAGGCTCTTTTTAAATGTATATCAGGCATGGCGGCGACTTGA
TAGTGAAAGTCCATATATGCTTTGTAGTCAAAACTGCTAGCGGATATTGTTATCTTAACA
...Header format:
NODE_1 is the number of the contig
length_251580 is the sequence length
cov_96.965763 is the k-mer coverage of the largest k used in assembly (will be discussed on Thursday)
Scaffolds are higher-order assemblies formed by ordering and orienting contigs
What do scaffolds indicate?
Paired-end reads provide distance and orientation information to connect contigs.
What is a scaffold FASTA file includes contigs linked by paired-end reads with "N"s as the base
Provides a higher-level view of genome assembly, bridging contigs to form scaffolds.
>NODE_1_length_335019_cov_108.862920
TTATATTGGCAGTAGTTGACTGAACGAAAATGCGCTTGTAACAAGCTTTTTTCAATTCTA
GTCAACCTTGCCGGGGTGGGACGACGAAATAAATTTTGCGAAAATATCATTTCTGTCCCA
CTCCCTAATTTAAACATTTTAAAATATACCAATTACTTTCATCCAAAGTGATCCTAAACC
AATCCAGATAATAAAGTAGACGAAACCTAATATTAAGTTCATTGTCCACCAACGTTTTTG
...
CATTTAAAATTTCTTGTGACATAGCATTCACCTCCTTTTAGAGCCACTTATTATTTATAA
TAATTAGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTGGCTCTTATGCA
GTTGGAGCGAAGATCCAACTGTAAACCATAGTGTACTTATTATTTATAATAATTAGTGGC
...
TTACTTTGAAATACTTTAAAAAAATAAGACACTTTCGTA
>NODE_2_length_262462_cov_97.035104>NODE_1_length_335019_cov_108.862920
TTATATTGGCAGTAGTTGACTGAACGAAAATGCGCTTGTAACAAGCTTTTTTCAATTCTA
GTCAACCTTGCCGGGGTGGGACGACGAAATAAATTTTGCGAAAATATCATTTCTGTCCCA
CTCCCTAATTTAAACATTTTAAAATATACCAATTACTTTCATCCAAAGTGATCCTAAACC
AATCCAGATAATAAAGTAGACGAAACCTAATATTAAGTTCATTGTCCACCAACGTTTTTG
...
>NODE_5_length_181792_cov_108.741524
TGGCTCTTATGCAGTTGGAGCGAAGATCCAACTGTAAACCATAGTGTACTTATTATTTAT
AATAATTAGTGGCTCTTATGCAGTTGGAGCGAAGATCCAACTGTAAACCATAGTGTACTT
ATTATTTATAATAATTAGTGGCTCTTATGCAGTTGGAGCGAAGATCCAACTGTAAACCAT
AGTGTACTTATTATTTGTAATAATATTGTAGAGTCTGAGACATAAATCAATGTTCAATGC
...Contigs
Scaffold
We almost always use this file for downstream processes.
Genome size (e.g., length of E. coli genome)
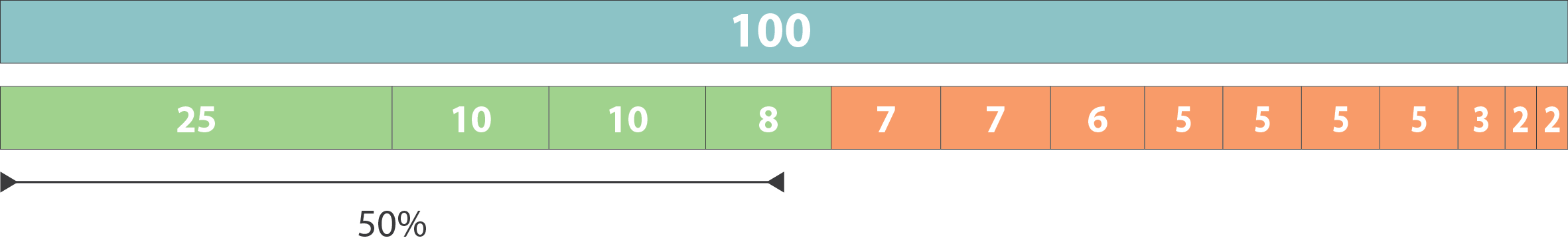
Higher N50 values indicate more contiguous assemblies
Largest contigs that make up the first 50
Remaining contigs
N50 = 8
Lower L50 values indicate fewer, larger contigs, which is better for assembly quality
L50 = 4
For L50, count the number of contigs used in the N50 calculation
Genome size (e.g., length of E. coli genome)
Largest contigs that make up the first 50
Remaining contigs
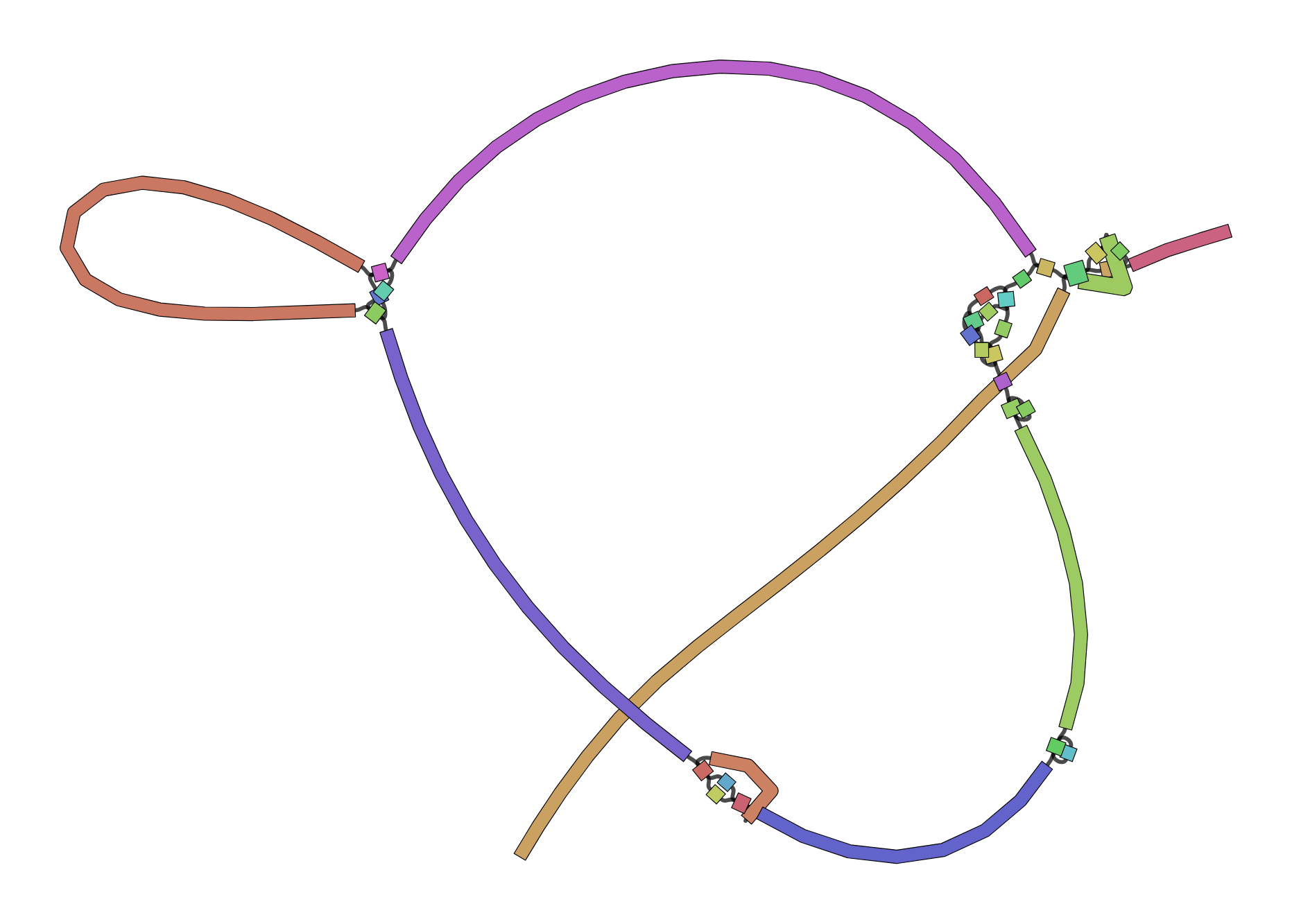
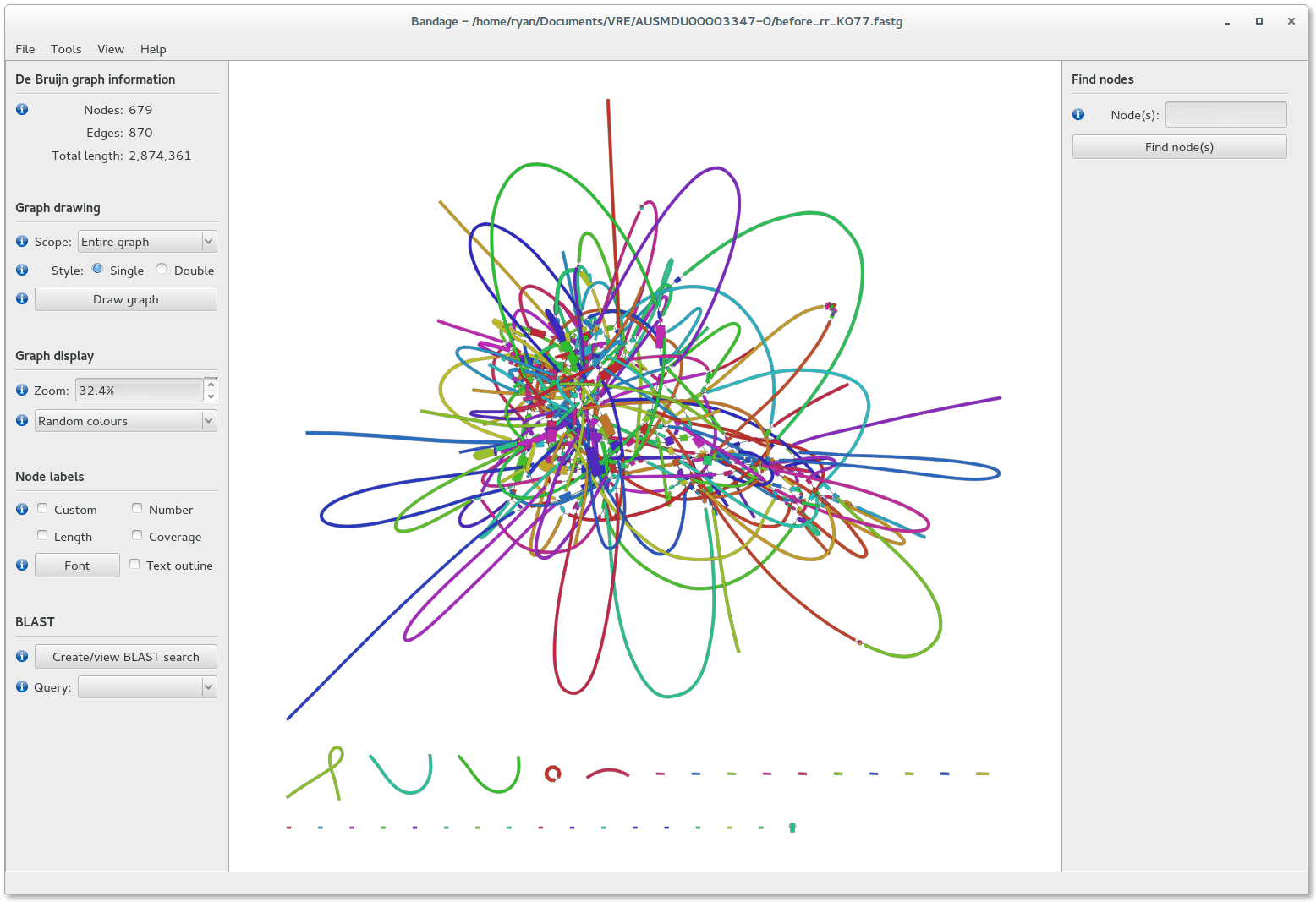
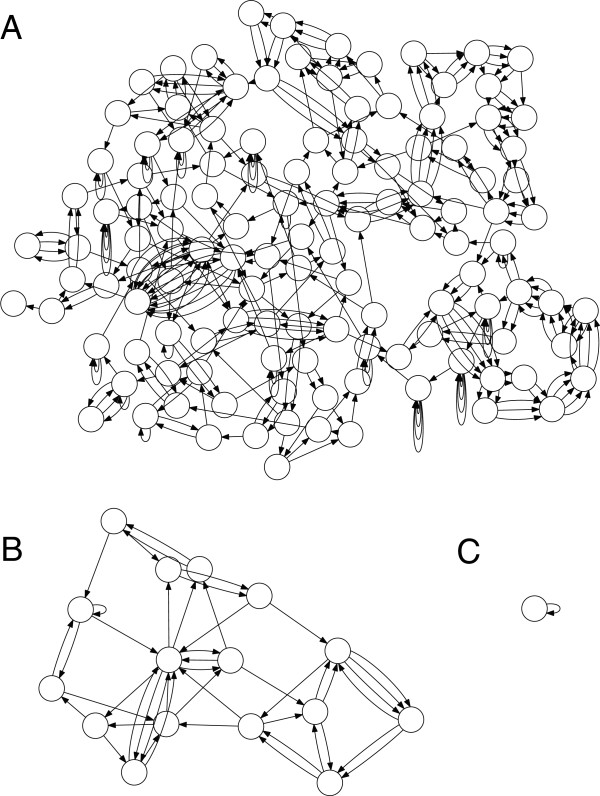
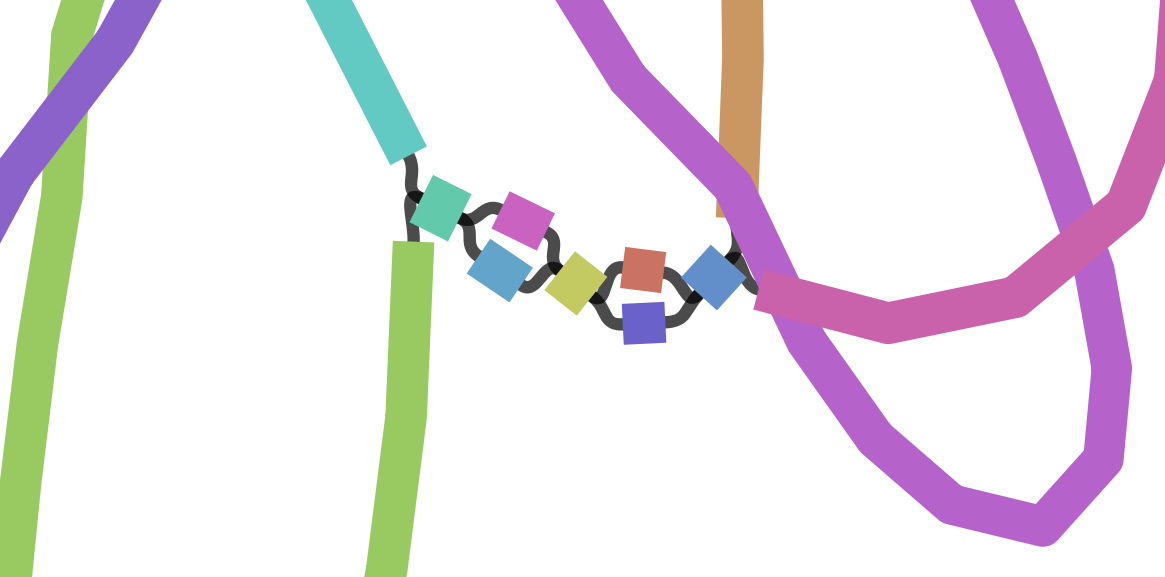
We can visualize contigs and how they connect with a Bandage graph
Each colored line is a contig/scaffold
Highly branched assemblies are not ideal
Here is an example of a real, highly branched assembly.
Islands are sequences that we cannot merge into our assembly above (often sequencing errors)
Lecture 03B:
Genome assembly -
Methodology
Lecture 03A:
Genome assembly -
Foundations
Today
Thursday
SPAdes is a popular prokaryote genome assembler
Based on De Bruijn graphs with numerous improvements
Leads to fragmented graphs and helps reduce repeat collapsing
Collapsed, tangled graphs great for low-coverage regions
By using multiple graphs, SPAdes can better handle variable coverage
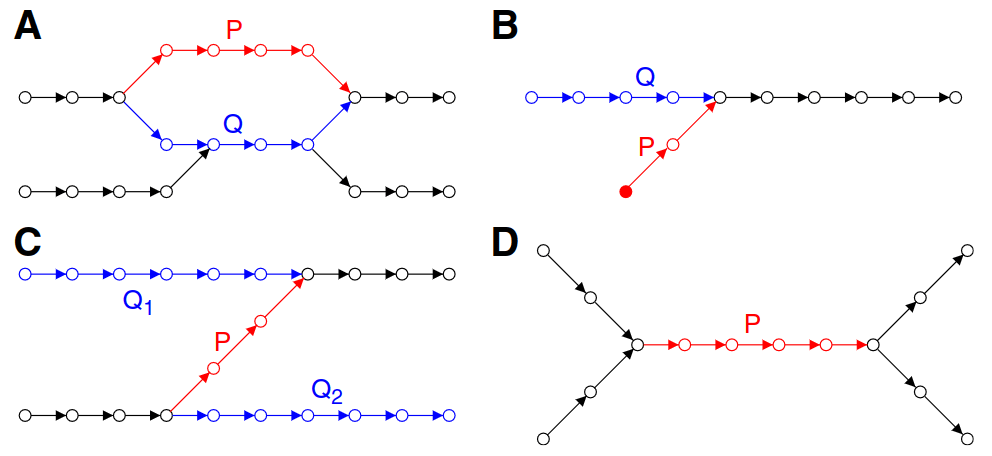
Large k
Small k
Potential bulge
Removal of a bulge will quickly deteriorate the graph and lose read information
If P needs to be removed, we "project" the information (e.g., coverage) onto Q
P's edges are then removed in the process
Potential tips
Removes P (shortest) and projects information onto Q
We can visualize this using an assembly graph from a tool called Bandage
Contigs
Scaffolds
Each island contains one or more contigs
Each solid line is called a "node" (Why? I have no idea.) and represent a contig
suggests how these contigs connect to form a scaffold
connection
Each