BIOSC 1540: L12A (Docking)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Apr 1, 2025
Lecture 12A
Docking
Foundations
Assignments
Quizzes
Final exam
OMETs
Proteins
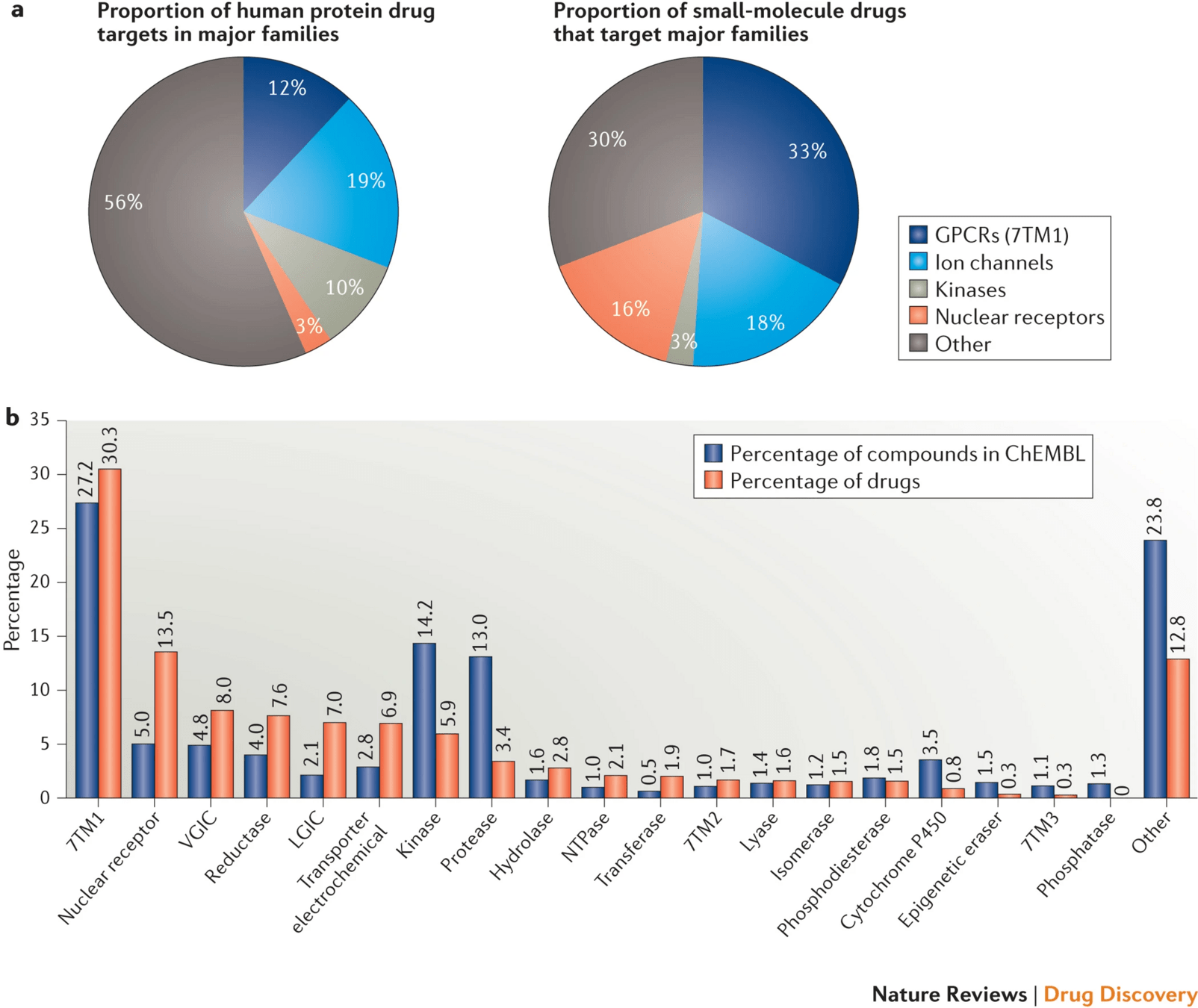
Over 90% of FDA-approved drugs act on proteins—enzymes, receptors, ion channels, and transporters.
These proteins play key roles in signaling, metabolism, immune response, and other vital functions.
Modulating protein activity with small molecules allows us to influence biological pathways precisely.
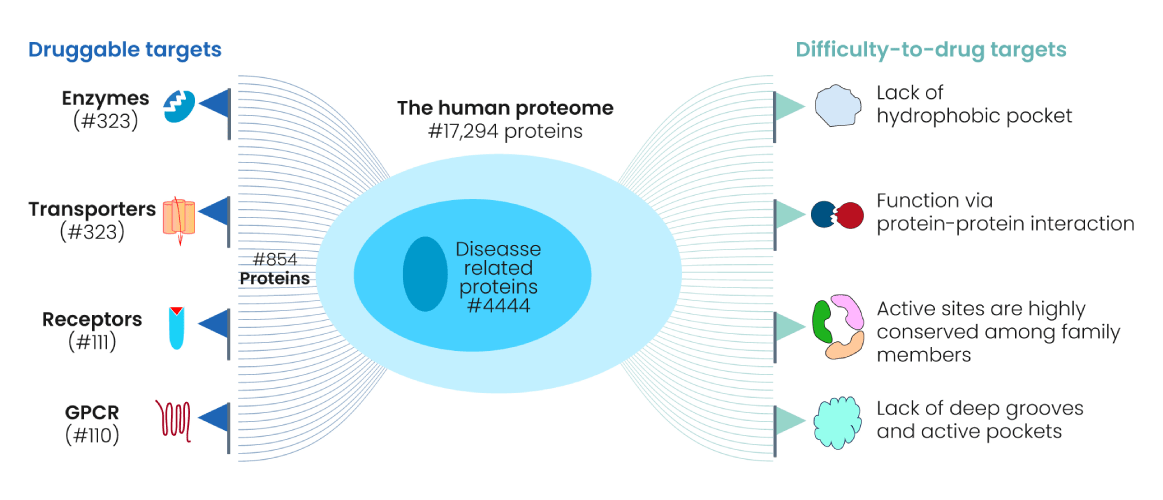
Not every protein is "druggable"—target selection must be biologically and chemically justified.
Genomics, proteomics, and phenotypic screens help identify candidate targets.
Criteria for Selecting a Protein Target
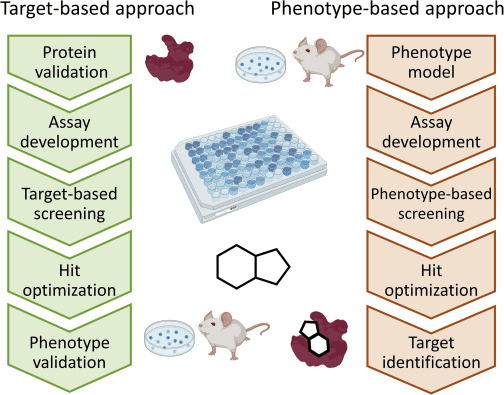
In target-centric approaches, researchers start with a well-understood protein and search for molecules that modulate its activity.
This contrasts with phenotypic screening, which starts with observed effects and works backward to find the target.
Target-centric methods allow rational design, structure-based modeling, and docking campaigns.
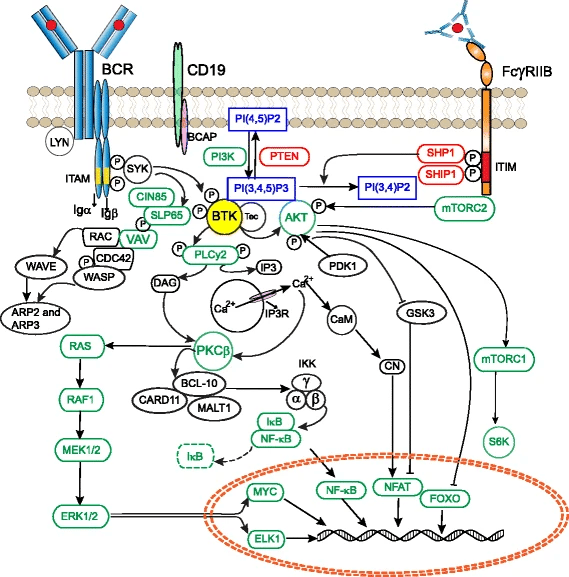
Bruton’s tyrosine kinase (BTK)
Proteins regulate nearly all cellular processes and drugs can inhibit or activate proteins to correct disease states
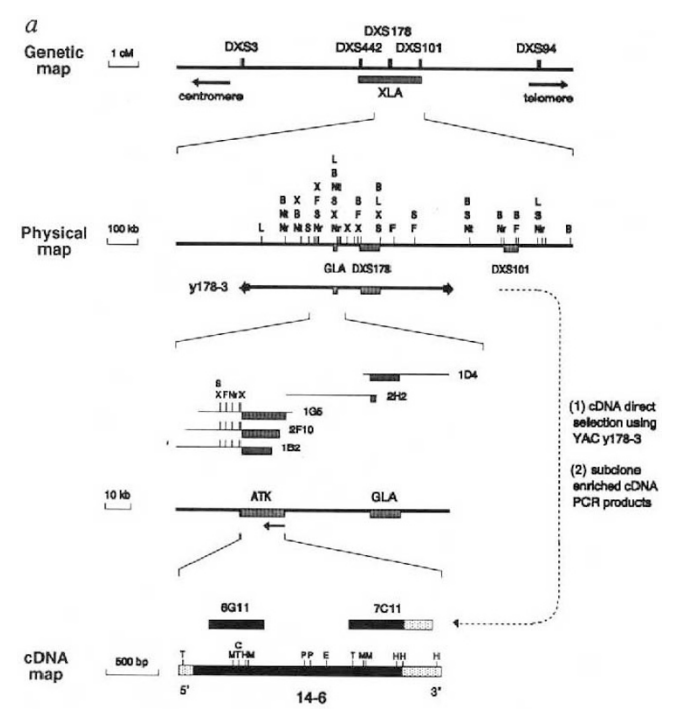
Example: Bruton’s tyrosine kinase (BTK) is a critical signaling enzyme that causes XLA, a genetic disorder marked by a severe lack of mature B cells that leads to immunodeficiency.
Family-based genotyping of affected males revealed tight linkage between the disease and genetic markers.
Bioinformatic comparison of the novel gene’s sequence to known kinases identified conserved domains with non-receptor tyrosine kinases.
In silico analysis of XLA patient sequences showed missense mutations affecting critical residues (e.g., Lys430 in the ATP-binding site).
Tyrosine phosphorylation datasets and known B-cell signaling proteins placed BTK at a convergence point of multiple BCR-related pathways
Comparative genomics shows high conservation of BTK’s domains across vertebrates that are overrepresented among central signaling hubs.
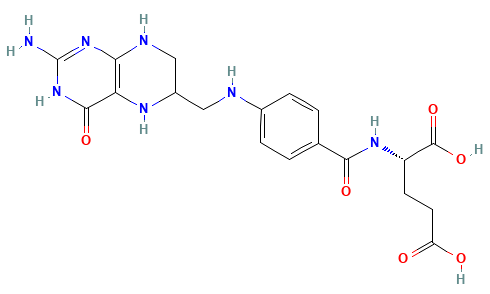
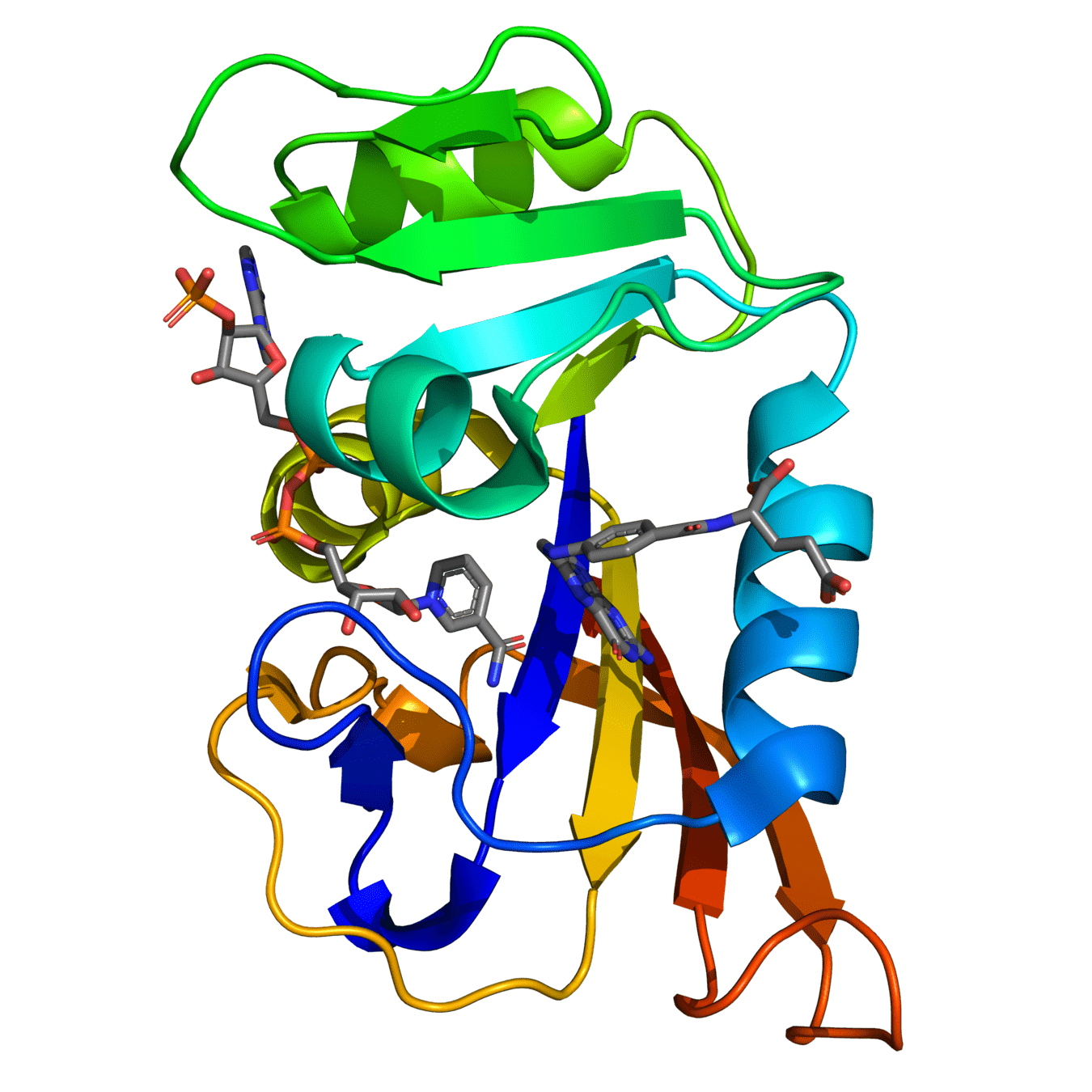
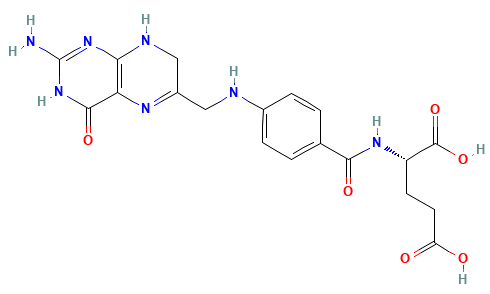
Dihydrofolate Reductase (DHFR)
THF is needed for
Disrupting THF production has a cascading effect on essential cellular processes, primarily affecting DNA and RNA synthesis and amino acid metabolism
This is a useful process for drug design
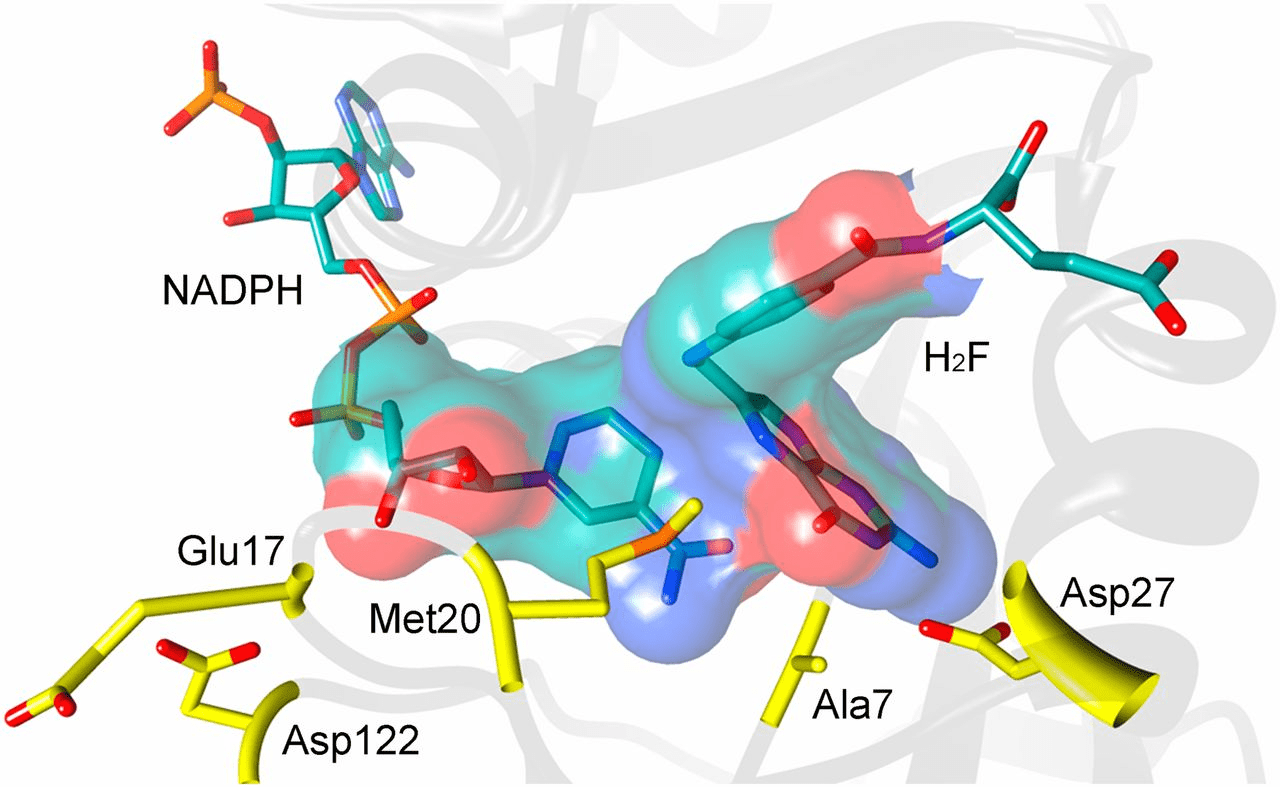
Dihydrofolate reductase (DHFR) is a crucial enzyme that produces THF from dihydrofolate (DHF)
DHF + NADPH
THF + NADP(+)
DHF
NADPH
(We will use this protein for our project)
DHFR has been extensively studied as an antibiotic (e.g., trimethoprim) and cancer (e.g., methotrexate) target
Patient could have deleterious side effects
What would happen if a patient with a bacterial infection is prescribed a drug loosely targeting DHFR?
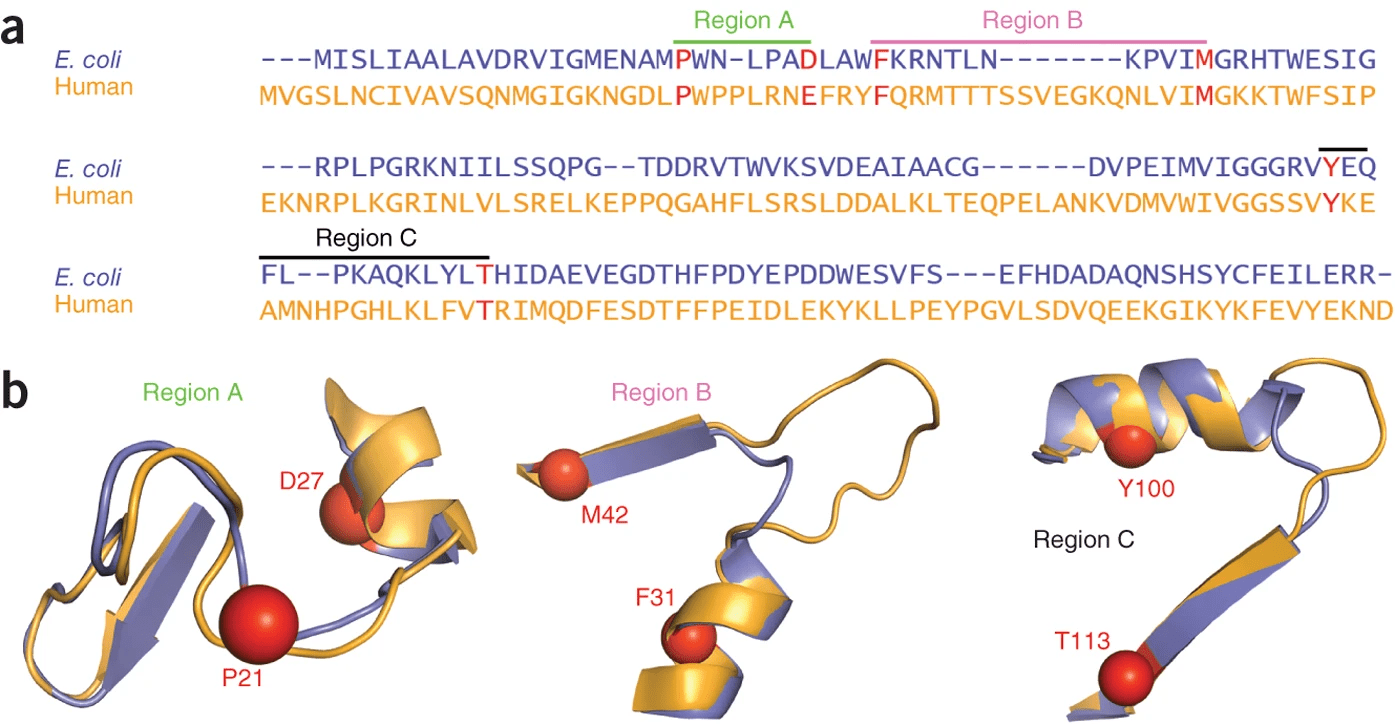
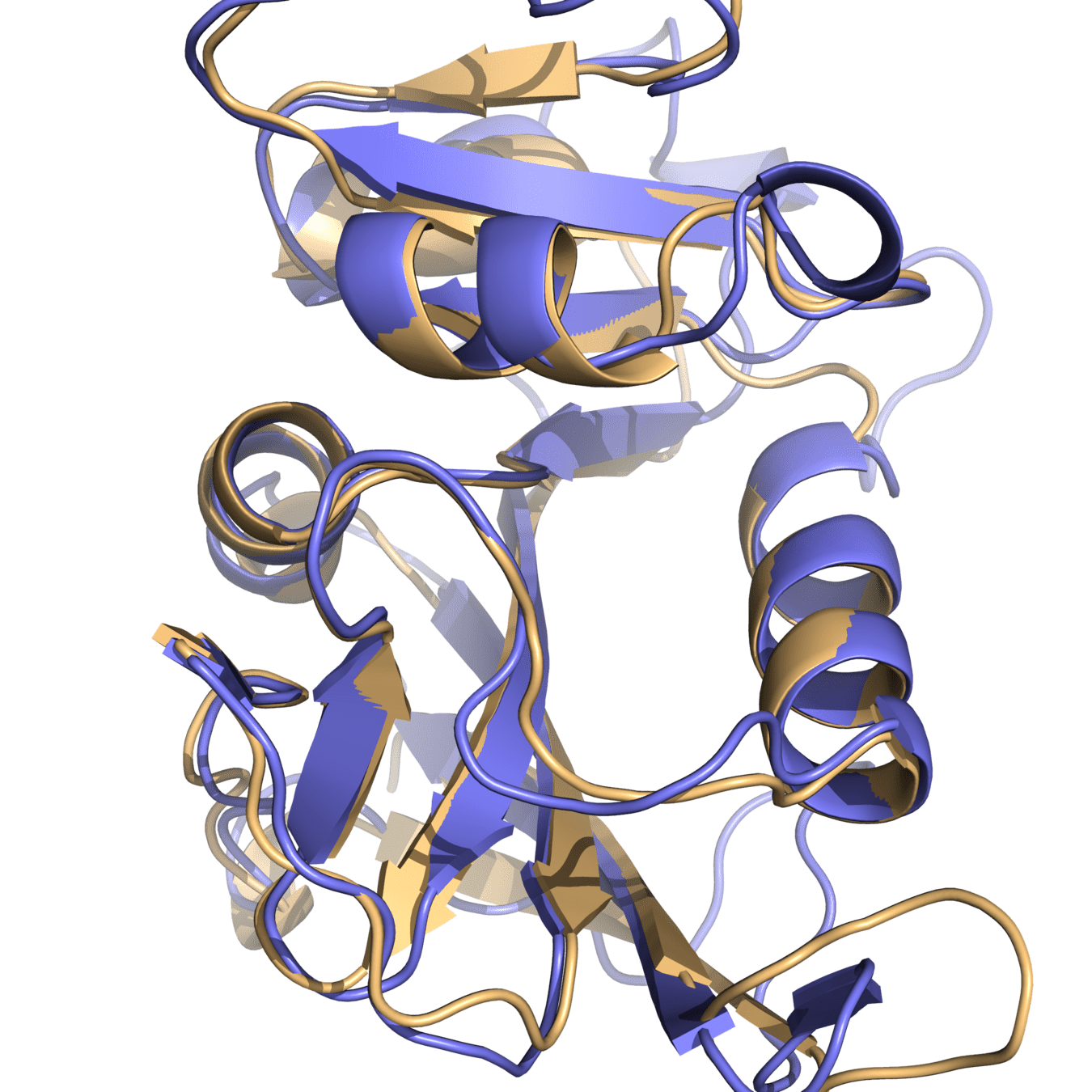
Both proteins have high structural similarity, even around the active site
Bacteria and humans have similar structures, but their dynamics are different
Outcome: We need to ensure drugs only bind to bacterial proteins by exploiting dynamic insights
A protein’s activity can be altered by binding a small molecule—often called a ligand—to a functional site on its surface.
This interaction can inhibit, activate, or subtly reshape the protein’s behavior.
These small molecules act like “molecular switches” that control protein action without altering the underlying gene.
Not every small molecule that binds a protein is useful—effective modulation depends on how tightly and selectively it binds.
High-affinity binding ensures that a drug is effective at low doses, while specificity minimizes off-target effects.
These properties can often be optimized through structure-based design and screening campaigns.
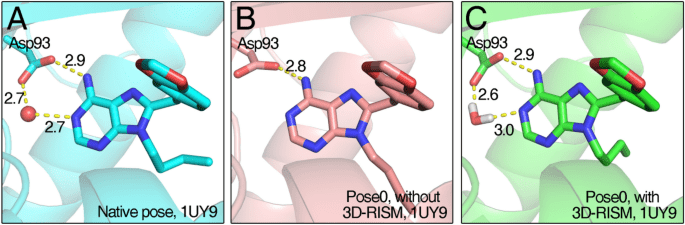
Critical ligand interactions in DHFR
Estimated to be between 1060 to 10200 possible small organic molecules
We need methods to navigate chemical space and identify promising leads accurately and efficiently
Experimental assays are still expensive, and limited to commercially available compounds
Instead, we can use computational methods to predict which compounds we should experimental validate
Can screen millions to billions of compounds in silico, thereby dramatically expanding our search space
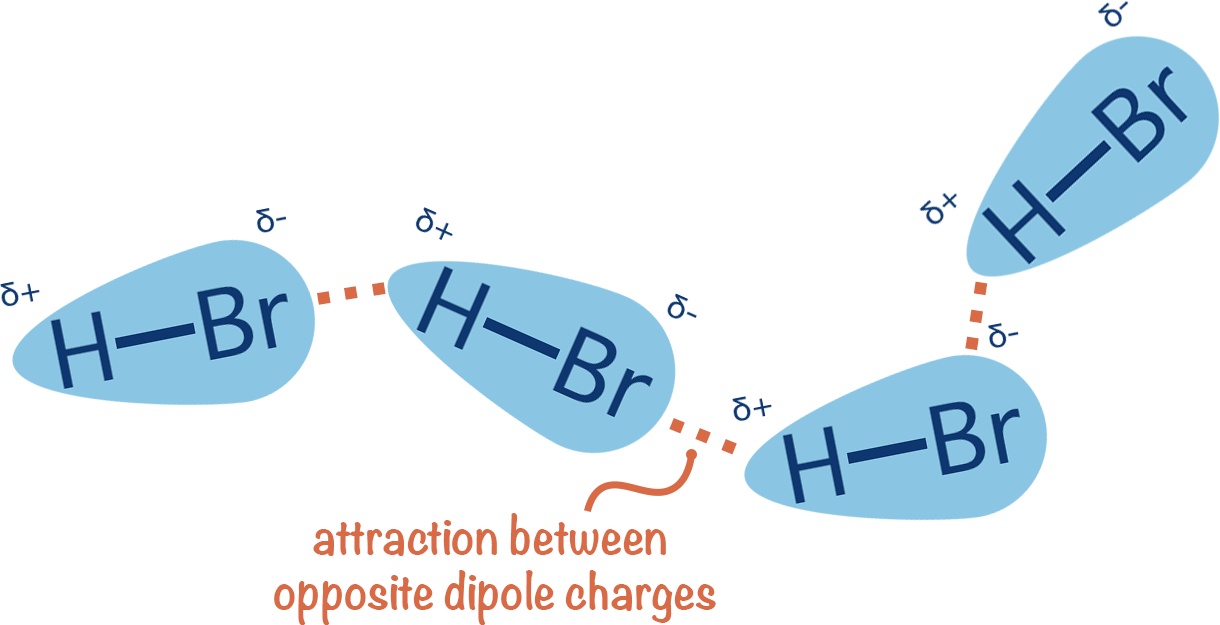
Binding occurs when a compound/ligand interacts specifically with a protein
Protein
Ligand
Binding
Protein-
ligand
We can model this as a reversible protein-ligand binding
Entropy
Enthalpy
Accounts for energetic interactions
How much conformational flexibility changes
By predicting the free energy of binding, we can identify small molecules with high affinity to our drug target
Objective: Directly predict binding affinity from protein and ligand structures with high accuracy and minimal computational resources.
We can carefully simplify our modeling to improve speed with (hopefully) minimal impact to accuracy
Avoid sampling all microstates and determine one "optimal" protein-ligand structure
Using this bound structure, predict a "score" that is correlated to binding affinity
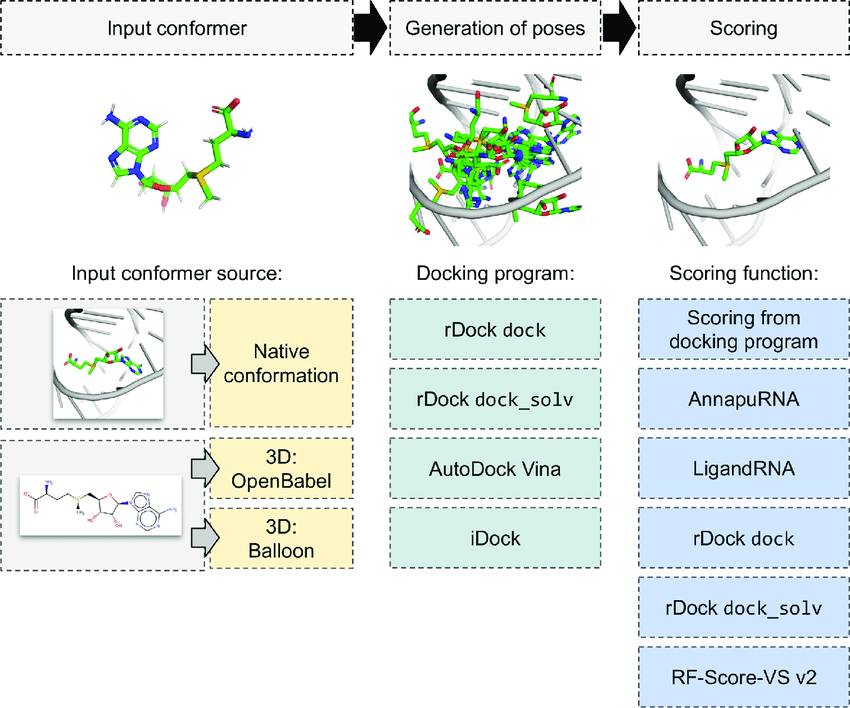
This is called docking
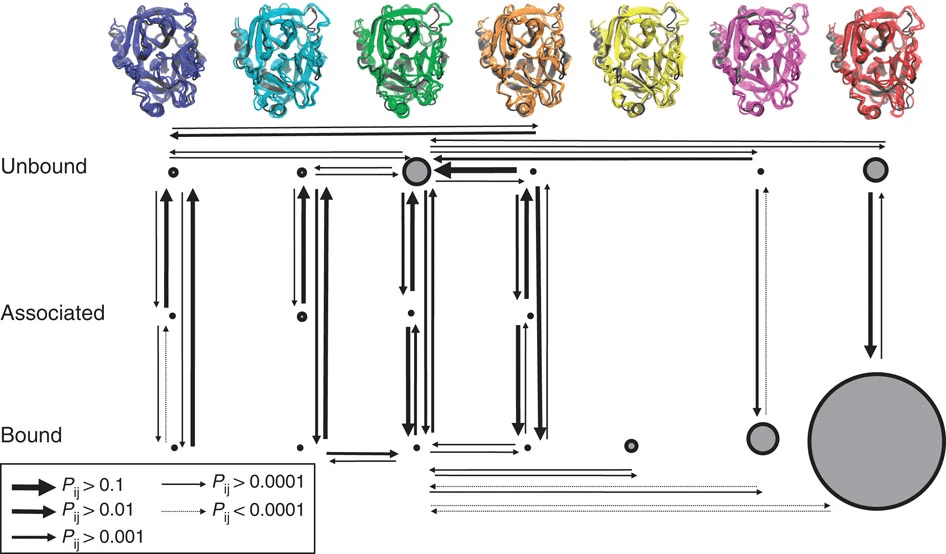
Significance of Protein Conformation in Docking
Docking still considers the protein structure, but we only select one
Experimental Methods
X-ray Crystallography: Provides high-resolution structures but may miss dynamic conformations.
Computational Techniques
NMR Spectroscopy: Captures ensembles of conformations but is limited to smaller proteins.
Will discuss these in L14
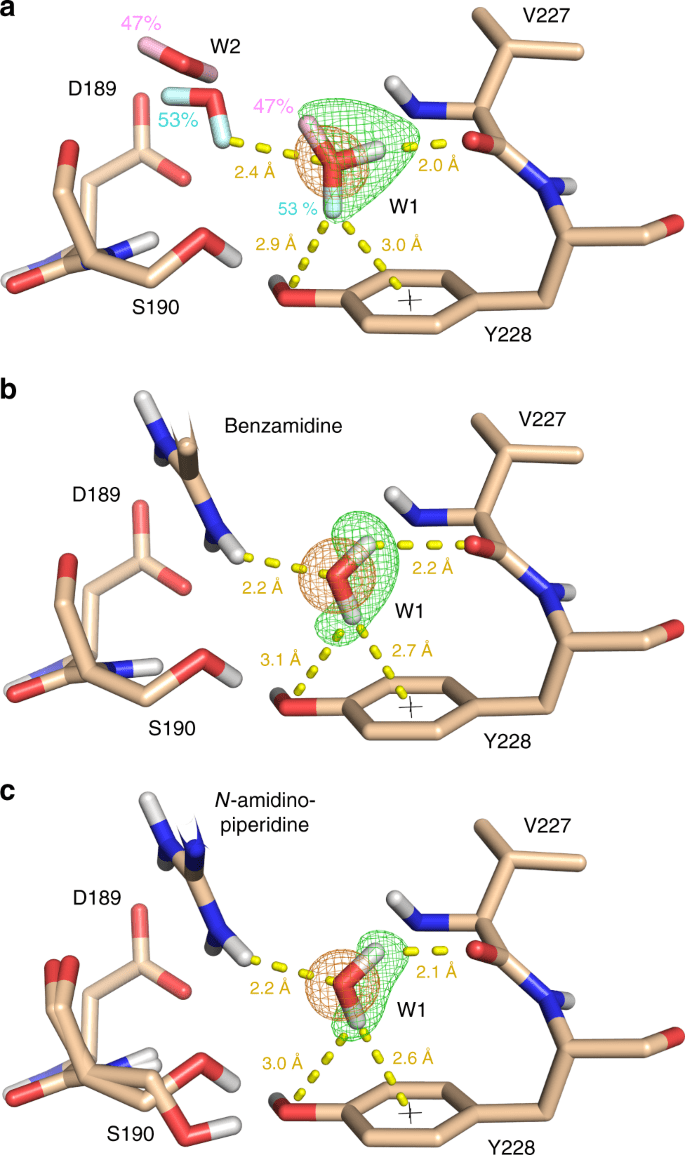
Role in Binding: Structured water molecules can mediate interactions between the protein and ligand.
Handling Water in Docking
Inclusion Criteria: Retain water molecules that are conserved across multiple crystal structures.
Types
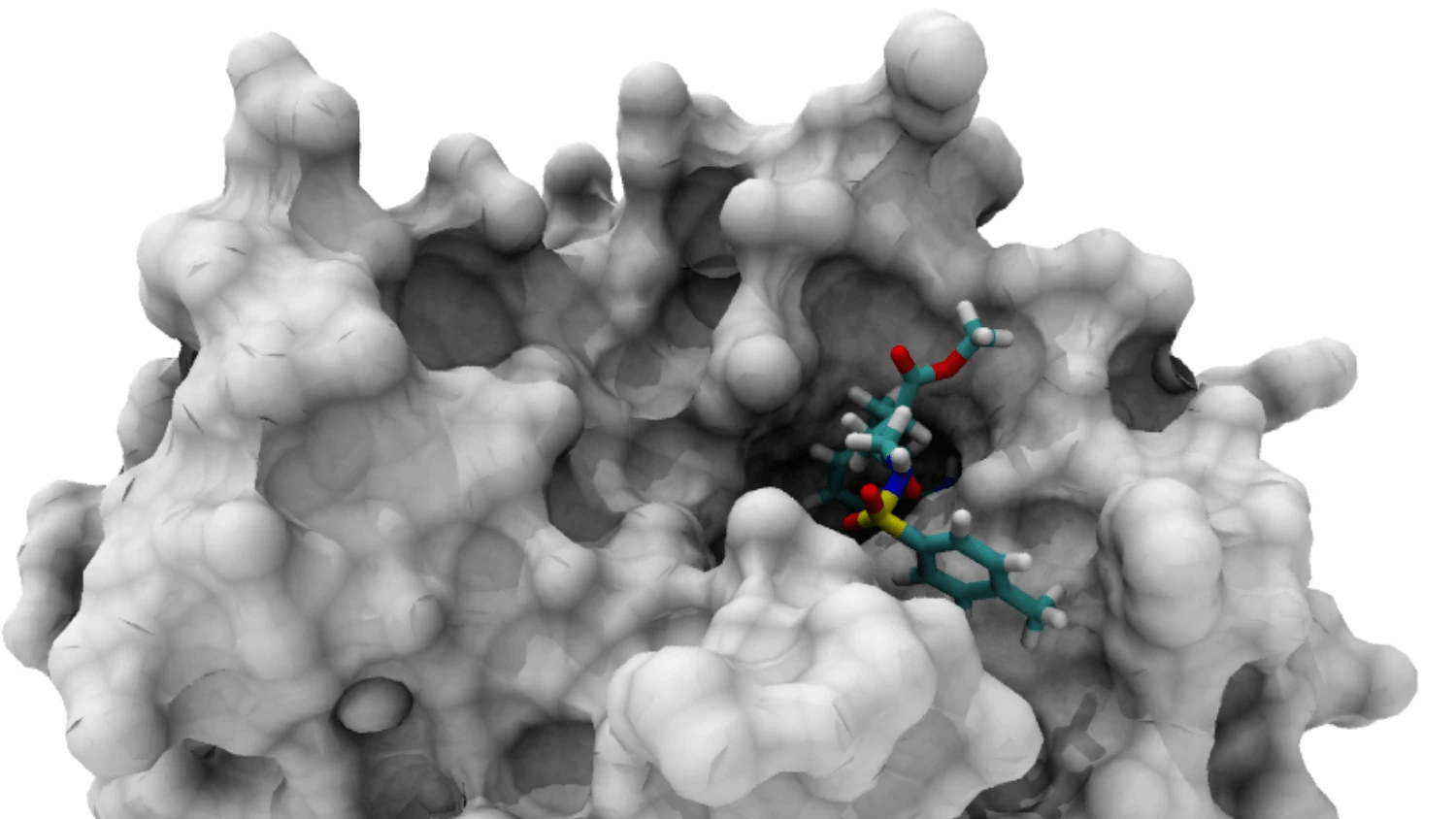
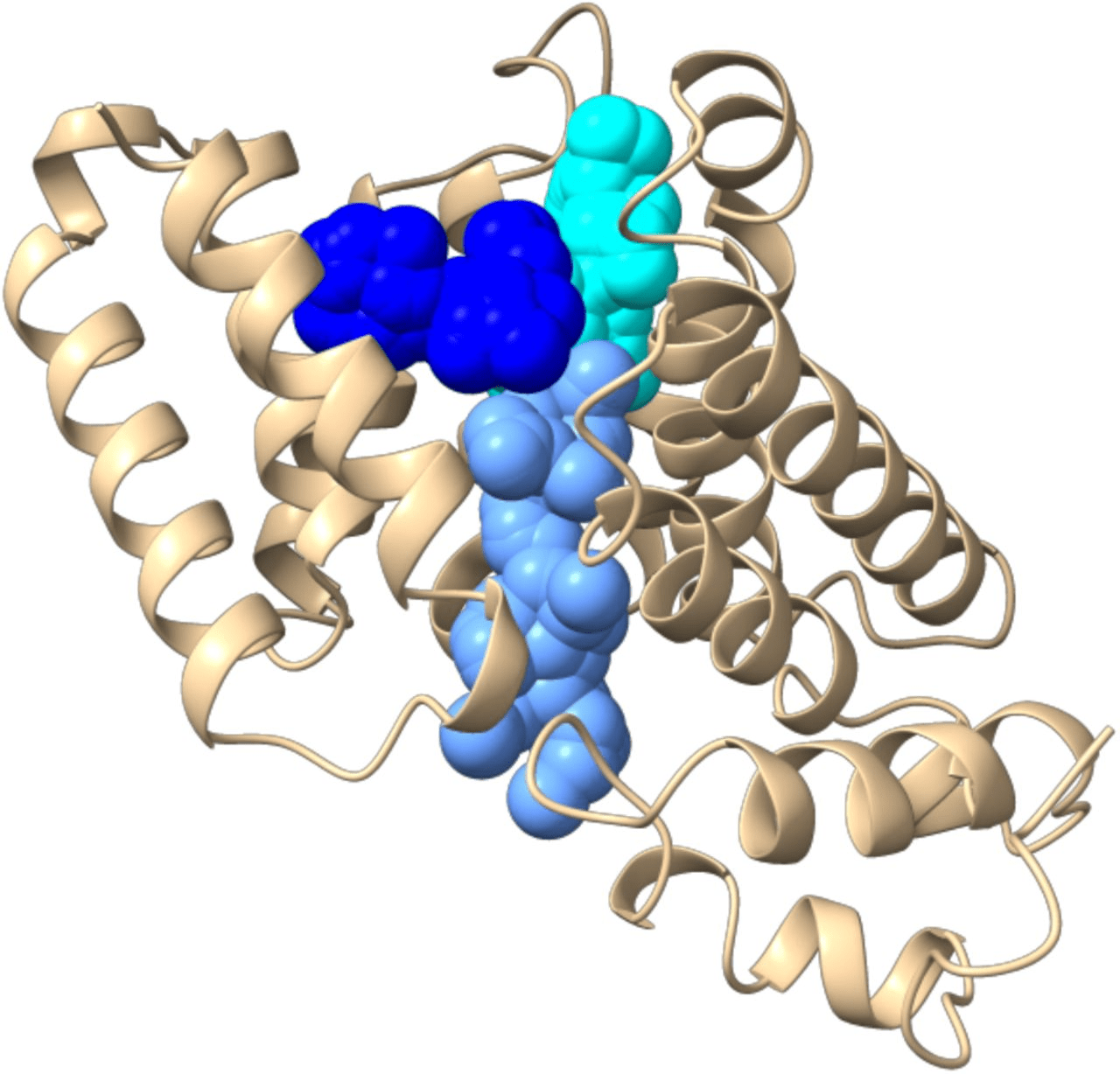
The binding pocket is the specific region where a ligand interacts with a protein
Accurate identification of binding pockets is essential for successful docking and virtual screening.
Active Site: The functional region where biochemical reactions occur (often a binding pocket in enzymes).
Protein Surface Characteristics
Binding Pocket: A cavity that can accommodate a ligand.
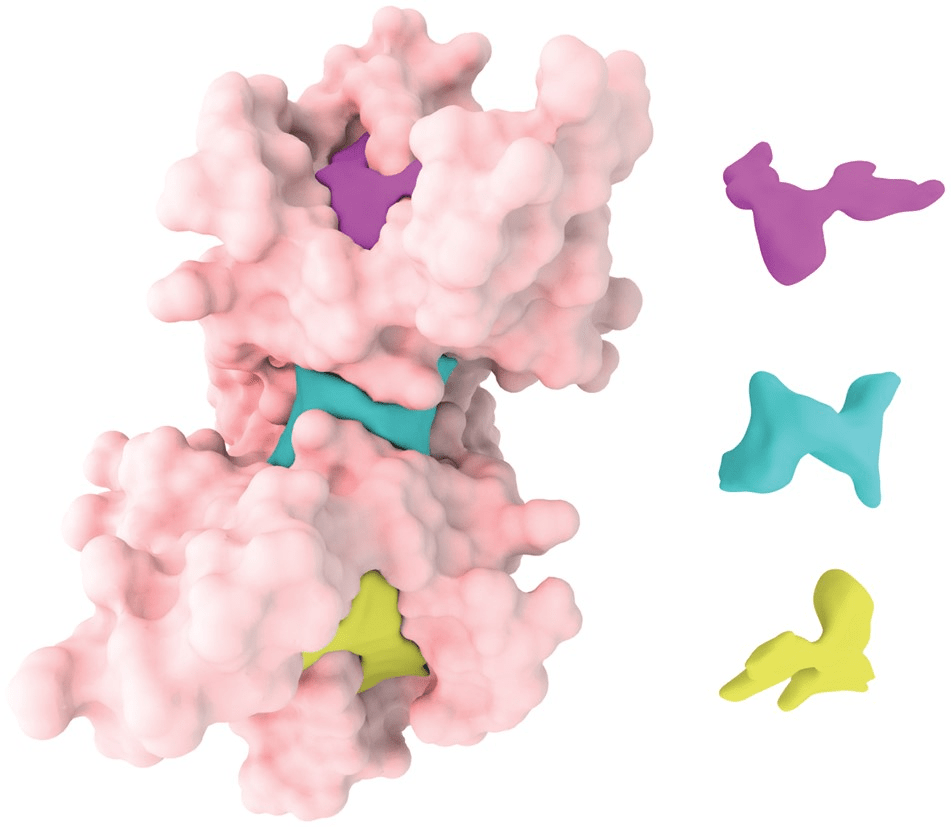
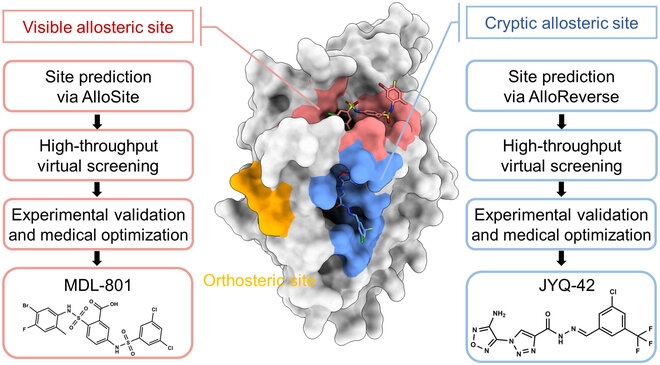
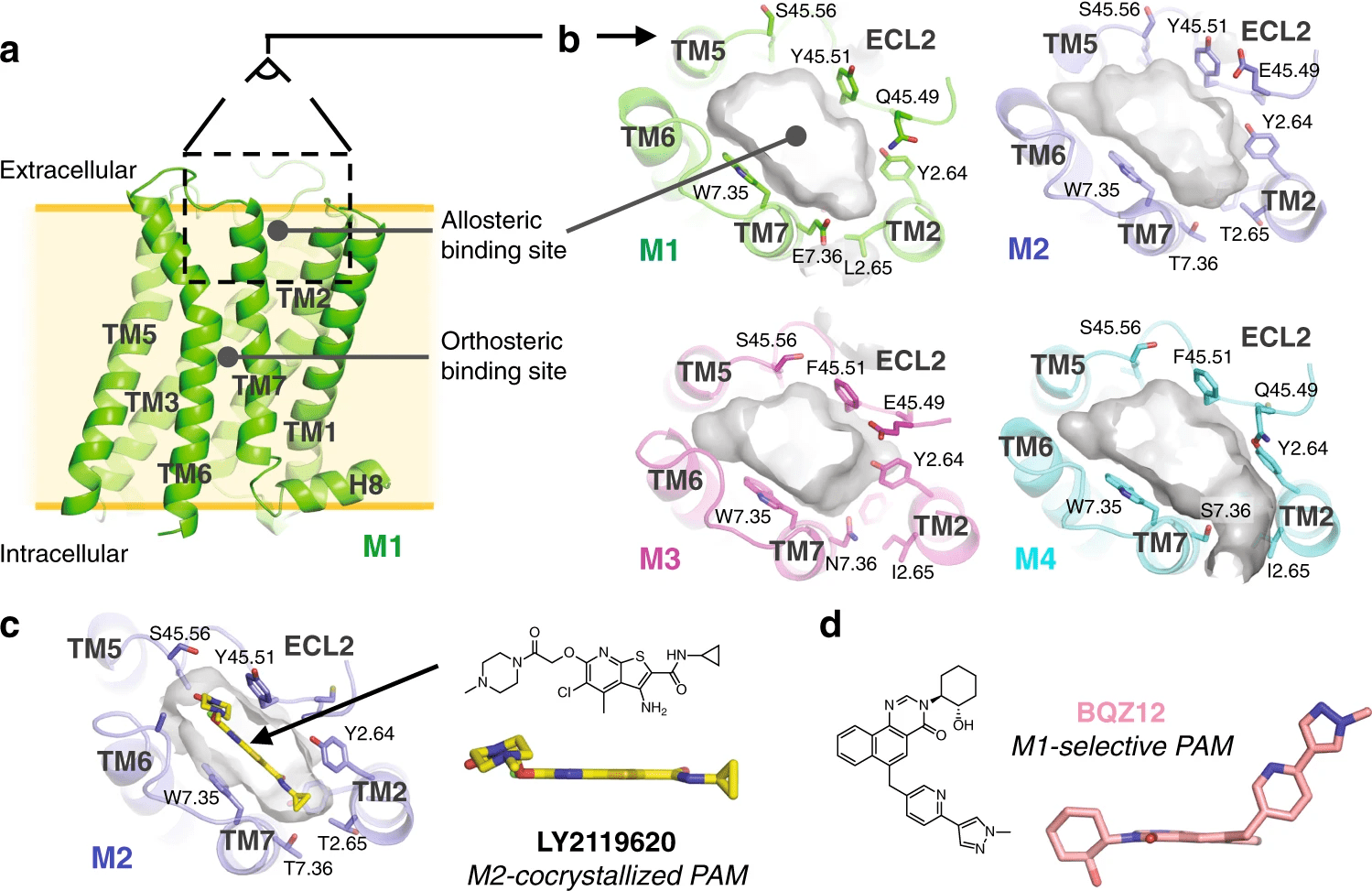
Orthosteric sites are the natural binding sites of endogenous ligands or substrates
Cryptic Sites: Binding pockets not apparent in the unbound protein structure but form upon ligand binding or conformational change.
Allosteric sites are spatially distinct from the orthosteric site and modulate protein activity indirectly.
Detection
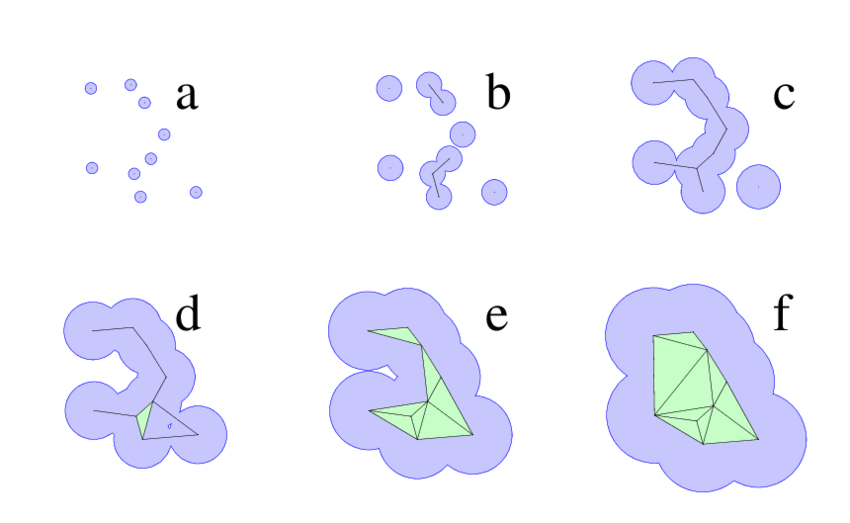
Alpha shapes extend the idea of a convex hull to capture the "shape" of a protein surface with cavities and tunnels.
Think of shrinking a sphere around atom centers—small alpha values allow more detailed surface features to be resolved.
Cavities enclosed by the alpha shape can be interpreted as binding pockets.
Proteins are modeled as a union of spheres (atoms), and the alpha shape filters through those to reveal cavities.
The algorithm identifies pocket volume, enclosure, and surface accessibility—critical for ligand fit.
These pockets are purely geometry-based, independent of electrostatics or residue type.
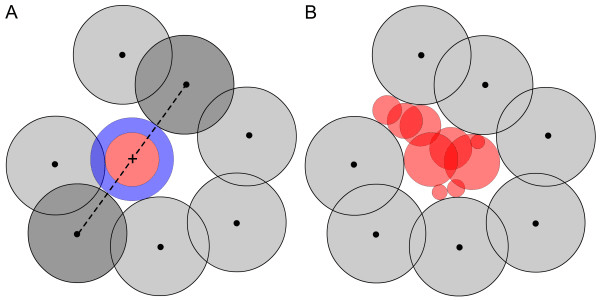
The protein is embedded in a 3D grid, and each voxel is labeled as protein, solvent, or cavity.
Grid points near the surface but not occupied by protein atoms are clustered into potential pockets.
Cryptic sites are hidden in the unbound structure and require conformational changes to become apparent.
Strategies
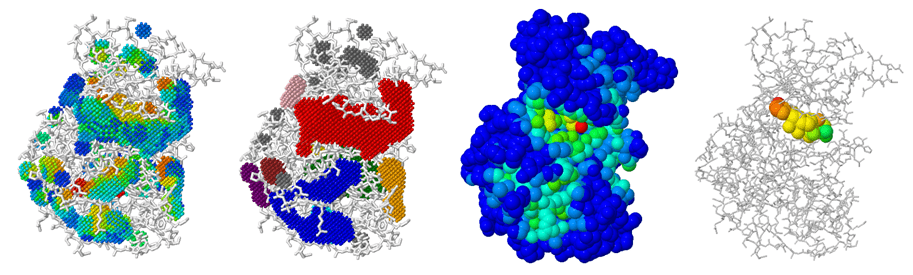
Case Study: Identification of allosteric sites in Hsp90
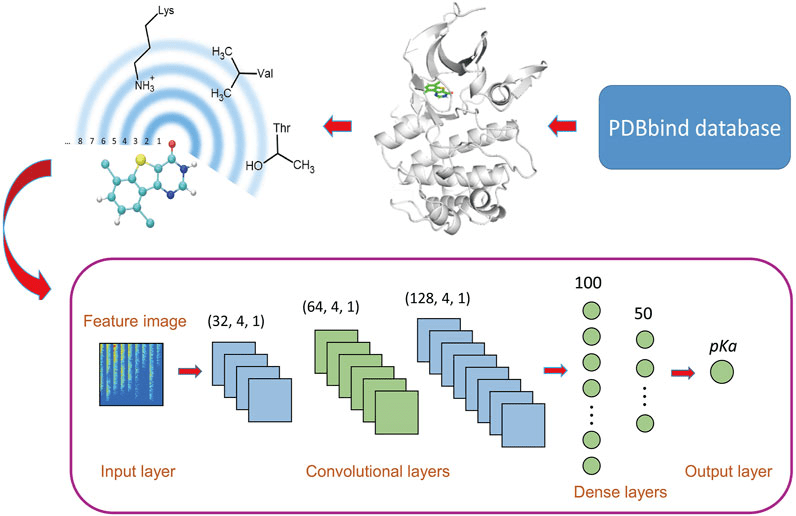
Scoring functions assign a numerical value to a protein-ligand pose, reflecting its favorability.
Scoring functions attempt to approximate this balance using simplified models.
The best pose maximizes favorable interactions (e.g., hydrogen bonds, hydrophobic packing) and minimizes clashes or strain.
A good scoring function may rank correctly even if absolute energies are wrong.
They consider van der Waals attraction/repulsion, electrostatics, and sometimes torsional strain.
Machine learning models (e.g., RF-Score, DeepDock) are trained directly on binding data.
These models learn nonlinear relationships and use structural and chemical features.
Require large, high-quality datasets and careful validation.
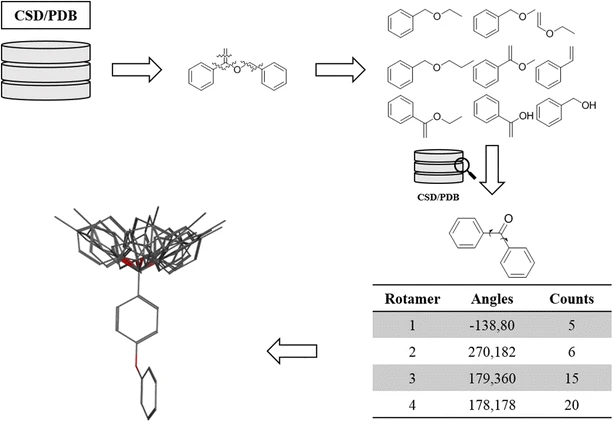
Search strategies
Identify important degrees of freedom
Scan along each angle with a step size of a N degrees
Remove structures with high strain
How many different conformations would we have in this molecule if we scanned only dihedrals every 45 degrees?
8 dihedrals
1
2
3
4
5
6
7
8
8 angles
8 × 8 × 8 × 8 × 8 × 8 × 8 × 8 = 16,777,216
That's a lot of structures, and many of them will clash!
We almost never do a systematic search in practice without some precautions to combinatorics
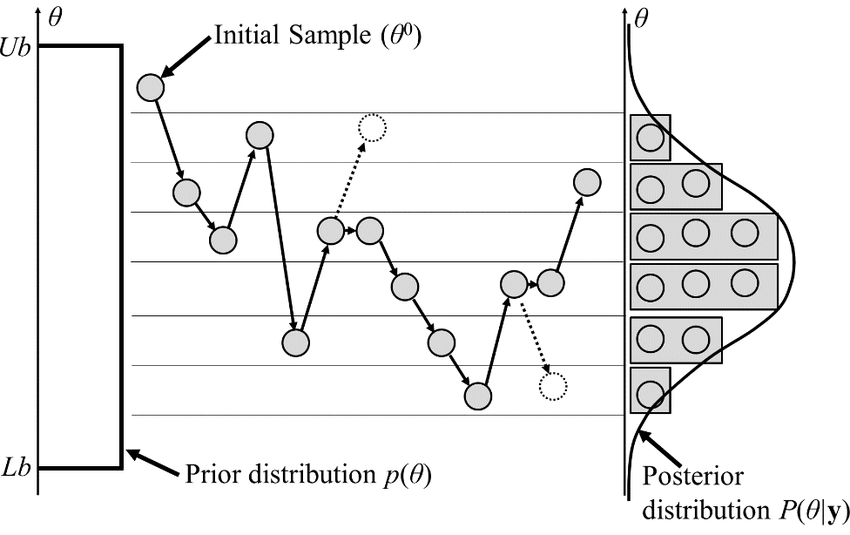
Monte Carlo
Steps:
Allows us to sample efficiently
Use pre-generated libraries
Lecture 12B:
Docking -
Methodology
Lecture 12A:
Docking -
Foundations
Today
Thursday