BIOSC 1540: L14A (Molecular simulations)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Oct 17, 2024
Lecture 13:
Molecular simulation principles
These motions are essential for binding, catalysis, and signal transduction
Understanding dynamics is crucial for drug design, protein design, biotech, etc.
Do not capture the full range of functional conformations
Simulation of Atomic Movements
Visualization and Analysis:
Refinement of Predicted Structures:
Studying Intrinsically Disordered Proteins:
Folding and Misfolding Pathways:
Quantum mechanics
Classical mechanics
Classical Mechanics
Quantum Mechanics
Nuclei
Electrons
Suitable Systems:
Limitations:
The acceleration of an object is directly proportional to the net force acting on it and inversely proportional to its mass
Given atomic forces, we can calculate atomic movements
Force vector acting on the particle [kcal/mol/Å]
Mass of the particle [amu]
Acceleration vector of the particle [Å/fs2]
How can we compute atomic forces?
The potential energy, U, is dependent on positions of all atoms
Forces are obtained from the negative gradient of potential energy
Determines acceleration and thus motion of atoms
Continuous motion approximated using discrete time steps
Time step length determines how "smooth" the animation/trajectory
Think claymation
3D coordinates of atoms in our system
These atoms exert forces on each other
Using Newton's equation of motion, we can predict their movement
Purpose of Integration Algorithms:
Challenges Addressed by Integration Algorithms:
Verlet: Uses current and previous positions to calculate the next position
Velocity Verlet: An extension of the Verlet algorithm that explicitly calculates velocities
Say we want to simulation system for 100 fs
Smaller time steps lead to more calcualtions to simulate same amount of time
0.5 fs
200
Number of total atomic force calculations
1.0 fs
100
2.0 fs
50
How can we do this quickly and accurately for large systems (e.g., proteins)?
GFN2-xTB; 1 fs time step; 1000 fs
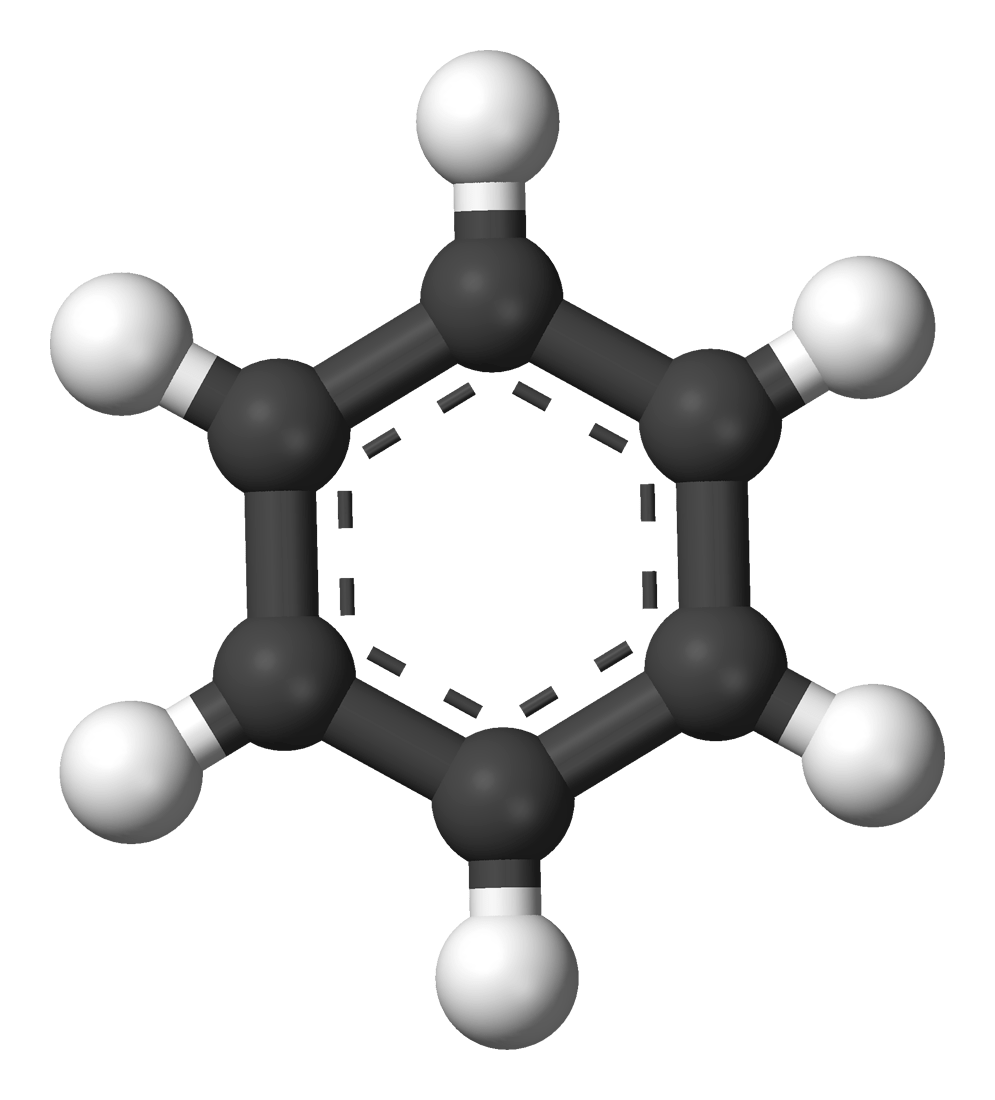
Let's consider a QM simulation a single methanol molecule (H3COH)
How would we model these atomistic dyanmics classically?
Bond lengths
Bond angles
Dihedral angles
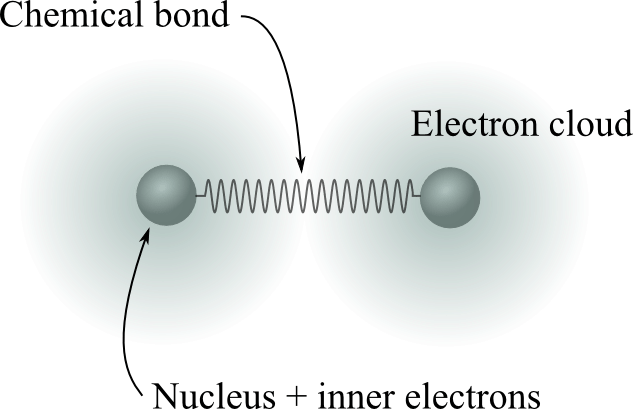
Two spheres (atoms) connected by a single spring
The spring resists changes in the distance between the two atoms
We can also get the force
Equilibrium bond length
Current bond length
Spring constant (i.e., bond stiffness)
CISD/cc-pVTZ
Single
Double
Triple
(These are approximate values.)
We can model each type of bond with a specific spring constant
We also have separate spring constants for bond angles
Three balls connected by two springs forming an angle, with a "hinge" at the central atom.
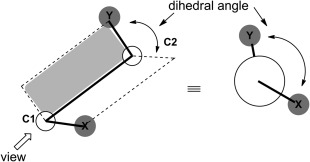
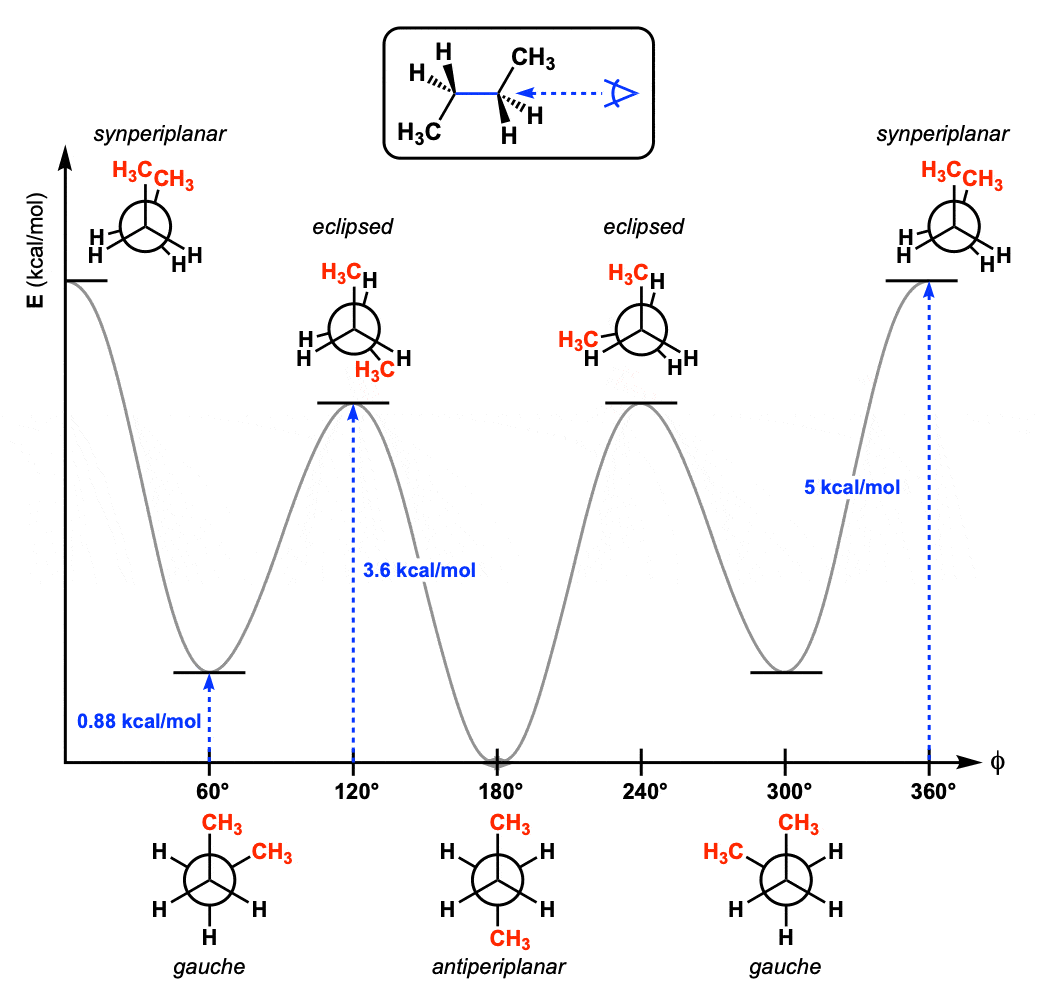
A dihedral angle is the angle between two planes formed by four sequentially bonded atoms (A–B–C–D)
It describes the rotation around the bond between atoms B and C
The dihedral angle 𝜙 is the angle between these two planes
Energies are computed with MP2/cc-pVTZ
Here, we have a periodic energy function with varying minima
How do we model this?
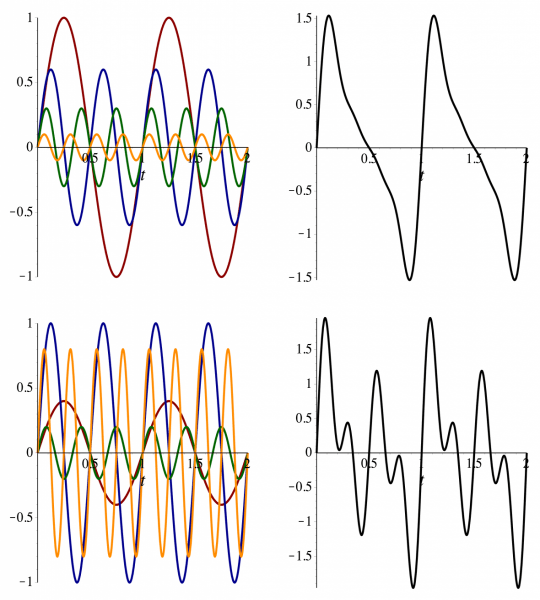
Constant term (i.e., average of function)
Amplitude coefficients
Harmonic number (i.e., frequency)
Higher harmonics add finer details to the approximation, enhancing the accuracy of the representation
Adding more sine and cosine terms improves the approximation, allowing the Fourier Series to closely match the original complex function
Dihedral angle
Amplitude for n-th Fourier
Phase shift
Number of periodic terms
Role in Molecular Assembly:
Facilitate the organization of molecules into complex structures.
Importance in Biological Systems:
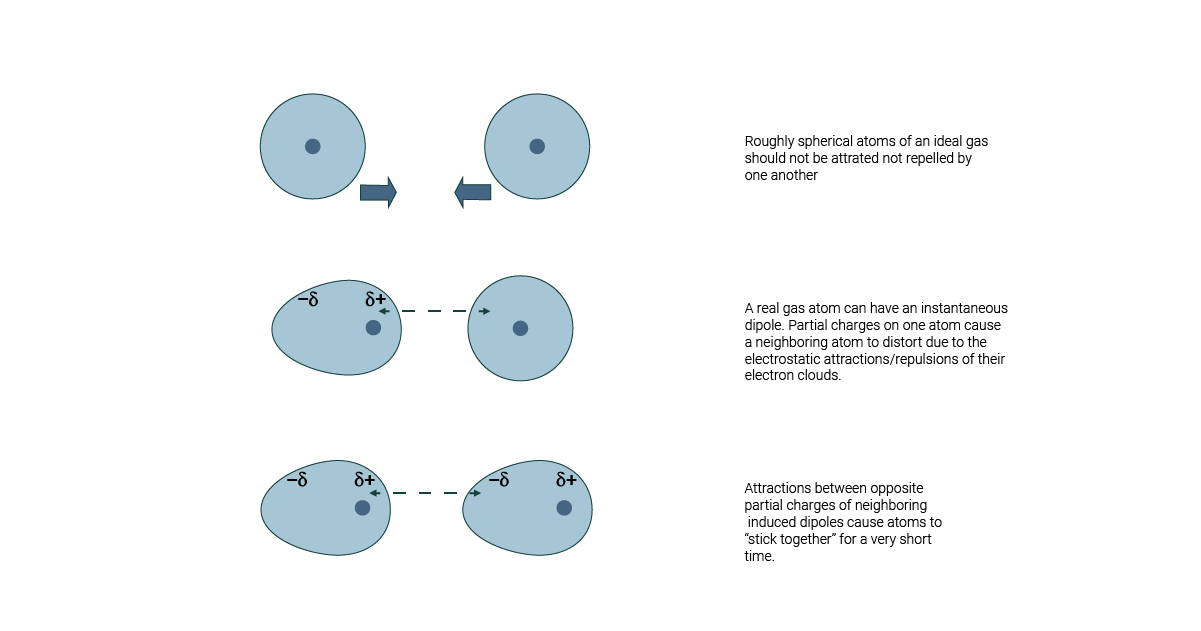
While covalent bonds define the primary structure of molecules, noncovalent interactions are pivotal in dictating how molecules interact
Dispersion coefficient
Repulsion coefficient
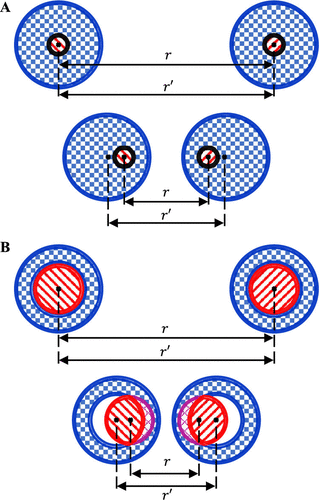
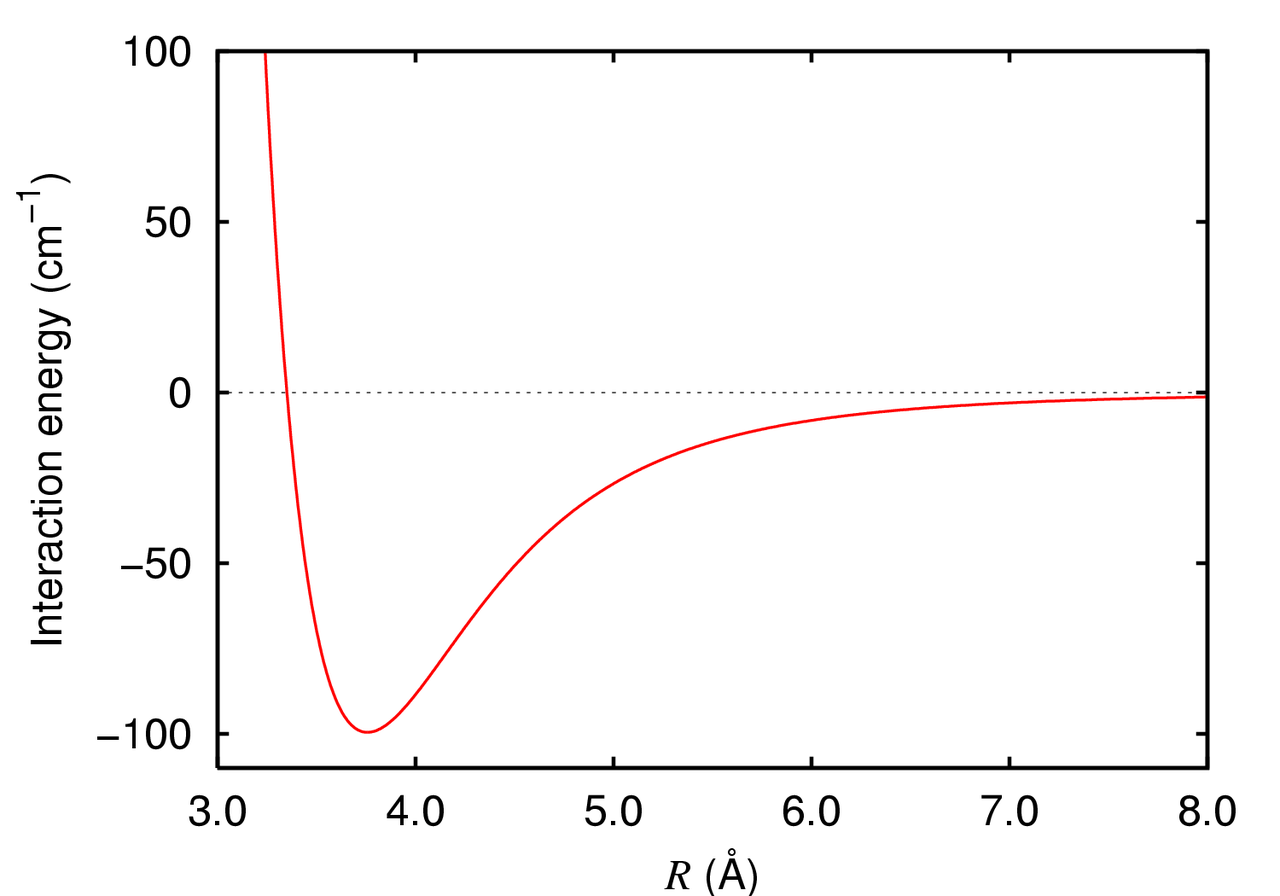
Van der Waals forces are modeled using the Lennard-Jones potential, which captures both the attractive and repulsive aspects of noncovalent interactions
Electrostatic forces decay as 1/r, making them significant over longer distances compared to van der Waals forces
Role of Quantum Mechanics:
Small Molecule Focus:
Complexity of Proteins:
Limitations of QM for Large Systems:
Types of Experimental Data:
Parameter Optimization:
Parameter Adjustment:
Iterative Refinement:
High Dimensionality:
Diverse Chemical Environments:
Dynamic Conformational Changes:
Long-Range Electrostatic Interactions:
Step-by-Step Process:
Common Force Fields:
Selection Criteria:
Limitations:
Lecture 13:
Molecular simulation princples
Today
Tuesday
Lecture 14:
Molecular system representations