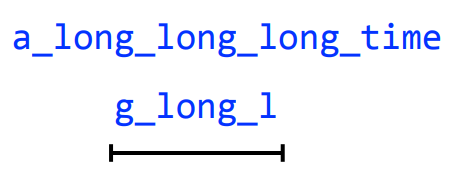BIOSC 1540: L03B (Genome assembly)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Jan 23, 2025
Lecture 03B
Genome assembly
Methodology
Assignments
Quizzes
CBytes
ATP until the next reward: 1,903
When asking for five FASTQ entries, here is what it should look like
fastq_five = """
@synthetic_read_1/f
TACGGCTAGGCATCTCGAGATCTGTGACGTTTCAGATCCCCTGCTGCGTGCGTTTGATGTCCAACTGTCGTACTCACGCCGGACGGGGAGTAACTTCTTTTCGAGCCGTAGTTGCGCGTACCCCATAAATTAGCCTTGGGAGCAGACAAGGCAAAACCCCAGAAGTTGGGTGTCCAATAAGGTTCACATCTGACCGGTCGCTTAAAAGGAAGCTTCCATTATCCGCTATATAAACGCCGGTGCTACCTAAGATGCGACACCT
+
46:47287653825380557902185865586;11784536:8>:7946436;67:04>8671293:53991474581727927476120866:4;;441889567264523394268:775;459726:6:5;865421764998273667561/6:6=987731855057787594555;4:859357388590:54:67365857865:66456:2737:3:34683758339648455674;6569;82364766;67
@synthetic_read_2/f
GACGATCGTAGCTCAGTCGGACCAACGACTCGCTGCTTACTGGAAGATCCTCGTAGACGGTTTTTTTGCGAAAGTACAGGCGACCCAGTACAAATCGGGATAGTGGTCACTTACTTATTTGGGCGGCTGCGATCATGGCCGCCGGTGGTTTAGGGTGTACGAATTCATCCTAGCCGCGAATGATTACGGACATCGTGACCATTATATGATCCGTTGTAGGCAGGTGTAACCCGACACTACTACATGACGGCTCACTCCAACA
+
GGDIHIFGEHGGIGGIHGFGIIFIHFDEFEFCCFFIIHIGIEEFIEFFICDGFHICFEICGGFFEIEEFGIFGFIHIIBDGHIGHIIGGGHFGIEHIIIDIIECAIHDHCEDECFHFBIIEIFHIIECIIIGIEIGEEFIDIHIEIFIDHFDIGEHEAGEIGDICIGFHHGGEIEDDDIGIDH?EIICDEHEHFGGIFGIGAIIIIIIIFGIEEIGCHIGIDIHIIIIICID@FIIIIEIGCIIGIHGIIIHIEGFHEEAFI
@synthetic_read_3/f
CGTAGCTGACGTAGATTCGATTTAAGAAACGCAGATATGGACATTGTCGCCGTGCCTTTATATTCCACATATCGTGGTAATCATACCGGCATAGGGTCATGTCCGCAGCTGTCCAACTATCGGTTAACGTTCCCCCTACTATCTCTGCGCGAGCCTAGAGTAAATCGATGAGTCTGAAGAACGCCTCATATCTGCTGTATGCCCGCCGCGTGAACTCTCAGTATTCGCGAACACATTGGTCTTGCTATCCTCGGTAAGGAAC
+
:=9<<:7<9::=<?<<6;;=;?;<7;9=9?6:8;8A9:=>=<:A79;=>=;:==:<4::7<9?E4<9;;:97=<7@9;8?@<7999:A9:=;6:?>:@988A?97=A>=@:;98>::<A>?7:<<A?<<5=7<7<==:4;3><=?5>><999;;=<8?;=88>>8;:;;7967>;;59=:<;8:7687856;679763<9=@;87550:627535<755461A329795;87269;45074317650467412984:51937
@synthetic_read_4/f
TACGGCTAGGCACGTTTTCAGCAATCACGCGTGAGAATGCAATACAGCTGAGTATAGGTGGCCGGGCGTACGTTTCTACGTGAGCATGTTTTTTTATTACAGAGTACCGGTAGCGGGGTTAACATGGAAGGCAGATACCGCTACGTCCGCCCCGAACGAGCCGCACATACAACCCGATCGGTACCGGTTAGTGTAAGGGGCTTTGTGAGTAGGTGTGGGCCGTGTACGGGTAGTACTCGGGAATGATGCGGATAATACCATC
+
>:A@=@=<ABB><=:==?>@=><<<9=?3:>@CHD;?=7:@?6G<8<@?AEE<=?;<;C<66B3>>>>=8488<8>?@9>43>?A?A61:@8;:6@97;825=>7>8><1<853;@B=66><A;?@5475:382@<@@<7><5-945?=7?;9=90<;B6C;;=77:?7;69-/87665=<<:>92A8AB47B5;8;3<A=8<92;7.99?65>653::@97A>6B>>11BAD>C>5:7,:A>@8<=7:<99??57:=:95<
@synthetic_read_5/f
GTACGATCGTACCTGCGTACAAAACAGTTTCGGGGTCCAAACCACGCCTCAACTGTTCTCGGTTAGTACCGTAGCTACACTCGGTCTATCTGTCAGCTGCCGTTCATTCGAGCTTTCGCGTACTTAACAGTACCAGAAGGGCGGGTCAGTATCCATGGTTGGTGCAAGCCCCTGTCCCAGTCTTACCGGATAGCGCACATTACTTCTCCTGGTTCGGGCGAATCCCCTAGCAATCCCATTTCATTACGAAACCAACGCTCCA
+
78<8675<68;9;9<72;4==:689<;95=5;?76:57<16;:4@;9.=:1:;?<49;89;0<>?6327778:8:518?7=79:6:<7><A@16:65<98:6<7446<;@9=9:<@<27:9<38@98<?8;9<5:4<3547?9>5:1;A=695=6<:8/;873?66::?45;:C857>:E:9:;9;258<:<<<79:599<<>77679<<;3<9328=53>8;35?E7=;8<A99>68;A99799?=5:6:70?<8::187;
"""Why we need genome assembly
DNA sequence (i.e., contig)
Overlapping reads
Assembly algorithms
TACGATCGGATTACGCGTAGGCTAGCTTACGGACTCGATGTACGATCGGATTACG
Recap from L03A
Assumptions
If we had two sources of DNA
Chance of overlap is likely, and it would be challenging to differentiate the origin of each read
+
It dramatically simplifies our problem if we assume only a single source of reads
TACGATCGGATTACGCGTAGGCTAGCTTACGGACTCGATGTACGATCGGATTACGCGTAGG
Real sequencing errors
can be fixed in high-coverage areas
Real SNPs
can be confidently detected when all reads have the same base
Assume that we have
high coverage
This happens all the time in science!
If you more robust options are available, using those may be required
If there is no other option, use the best approach and disclose how this could impact your results and interpretation
String manipulation in Python
read1 = "ATCG"
read2 = "TCGA"
A DNA sequence is simply a sequence of letters: A, T, C, and G. In Python, we can represent this using quotation marks ("" or '').
read_long = """
CGTAGCTGACGTAGATTCGATTTAAGAAACGCAGATATGGACATTGTCGCCGTGCCTTTATAT
TCCACATATCGTGGTAATCATACCGGCATAGGGTCATGTCCGCAGCTGTCCAACTATCGGTTA
ACGTTCCCCCTACTATCTCTGCGCGAGCCTAGAGTAAATCGATGAGTCTGAAGAACGCCTCAT
ATCTGCTGTATGCCCGCCGCGTGAACTCTCAGTATTCGCGAACACATTGGTCTTGCTATCCTC
GGTAAGGAAC
"""
read1 = "ATCG"
read2 = "TCGA"
# Compare two strings
print(read1 == read2) # Output: False
To compare strings, we can use the equality operator ==
read1 = "ATCG"
read2 = "ATCG"
# Compare two strings
print(read1 == read2) # Output: True
0123
ATCGUse square brackets [] to get a character by its index
read = "ATCG"
print(read[0]) # Output: A
print(read[2]) # Output: C
Use slicing with start:stop to get part of a string
print(read[0:2]) # Output: AT
print(read[1:3]) # Output: TCPython does not include the stop index
Each character in a string has an index
Example: "C" has index of 2
Use a for loop to go through each character one by one
read = "ATCG"
for char in read:
print(char)
# Output:
# A
# T
# C
# G
read = "ATCG"
for i in range(len(read)):
# Print substrings starting at index i
print(read[i:])
# Output:
# ATCG
# TCG
# CG
# G
You can also slice inside of a for loop with an index
range(len(read))generates integers from 0 until the length of the read (in this case 4)
read1 = "ATCG"
read2 = "TCGA"
for i in range(len(read1)):
if read1[i:] == read2[:len(read1) - i]:
print(f"Overlap found: {read1[i:]}")
break
# Output: Overlap found: TCG
Let’s find where read1 overlaps with read2
When i = 0:
read1[0:] gives us "ATCG" (the full string)read2[:4] gives us "TCGA" (first 4 characters)"ATCG" == "TCGA"
read1 = "ATCG"
read2 = "TCGA"
for i in range(len(read1)):
if read1[i:] == read2[:len(read1) - i]:
print(f"Overlap found: {read1[i:]}")
break
# Output: Overlap found: TCG
Next is i = 1:
read1[1:] gives us "TCG" (excluding 'A')read2[:3] gives us "TCG" (first 3 characters)"TCG" == "TCG"
Once we find the overlap, we can merge the reads
read1 = "ATCG"
read2 = "TCGA"
i = 1
merged = read1[:i] + read2
print(merged)
# Output: ATCGA
We can use this approach of finding overlaps and merging reads to form a contig
This idea of finding overlaps and merging motivates our first assembly approach: the greedy algorithm
Overlaps and merges
Algorithm
1. Check every possible read for the largest overlap.
2. Merge the two reads with largest overlap.
3. Repeat until no further merges are possible.
At the end, we have a set of contigs that represent our original DNA sequence
The greedy algorithm focuses on selecting the best immediate option (i.e., local optimal) at each step without full consideration of the overall global solution
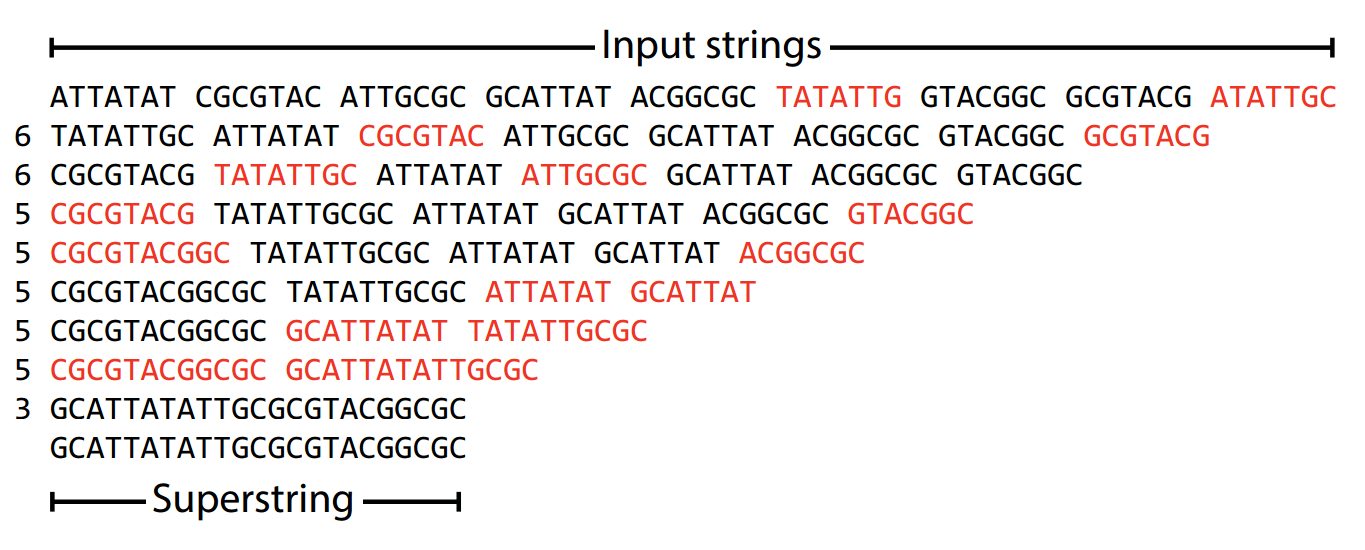
The greedy algorithm aims to find the shortest superstring, which minimizes unnecessary duplication.
A superstring is a single string that contains all reads as substrings
Example: ACGTAC is a superstring of
ACGT, CGTA, GTAC
This means the greedy algorithm will always make the best move in the moment even if it gives the wrong final answer
Breaking ties
Talk with your neighbors
Suppose we have these three reads
ACGTAA
CGTAAC
with a highest
overlap of five
We merge reads R2 and R3:
TAACGT
R1
R2
R3
TAACGT
ACGTAAC
(and keep R1)
However, now we have a problem
ACGTAACGT
TAACGTAAC
Both have a length of 9, which one is the correct move?
Overlap of 4
TAACGT
ACGTAAC
Overlap of 4
TAACGT
ACGTAAC
First encountered, first merged
Highest quality base calls
Highest coverage
Look ahead
Exclude
The one you found first
Use sequence with highest quality
Whichever results in more coverage
Do both and evaluate consequences
Be petty and don't merge them (separate contigs)
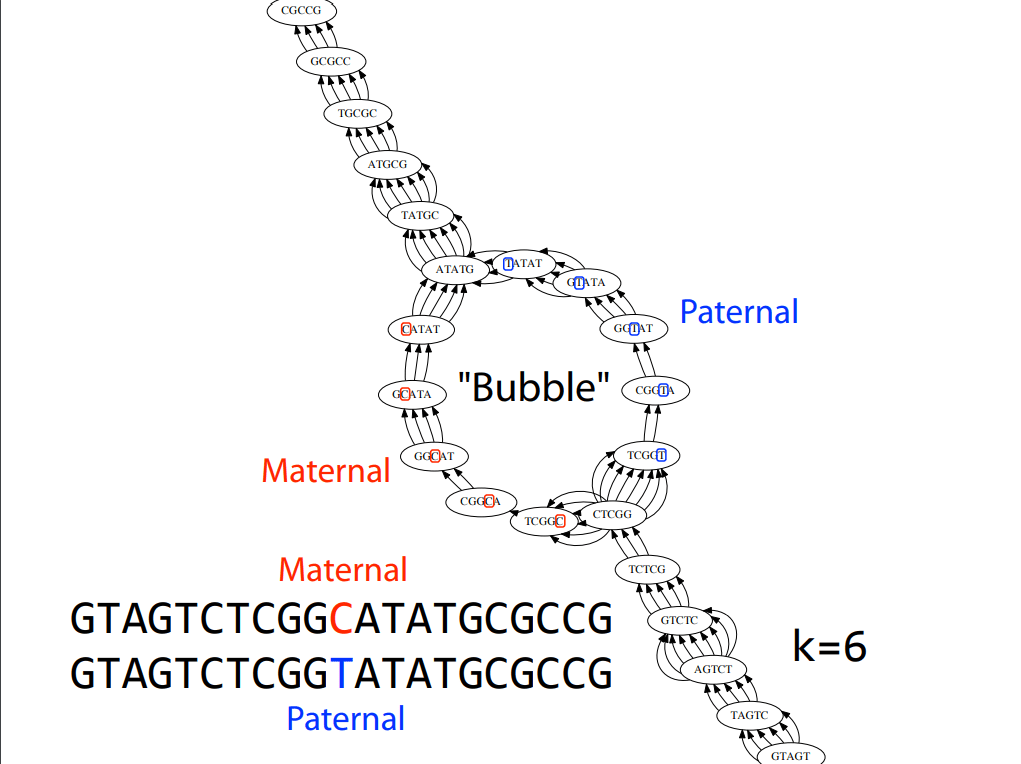
Trouble with repeats
a_long_long_long_time
We are missing a "_long". Why?
Let's take a string and cyclically permute it with k = 6
Then perform the greedy algorithm
We get the correct string back, but how did increasing our k fix this?
a_long_long_long_time
By having one read span all three "long"s, (i.e., the repeating region) we prevented a collapse
k = 8
Remember: This is why long sequencing reads are very helpful in resolving repeats!
K-mers
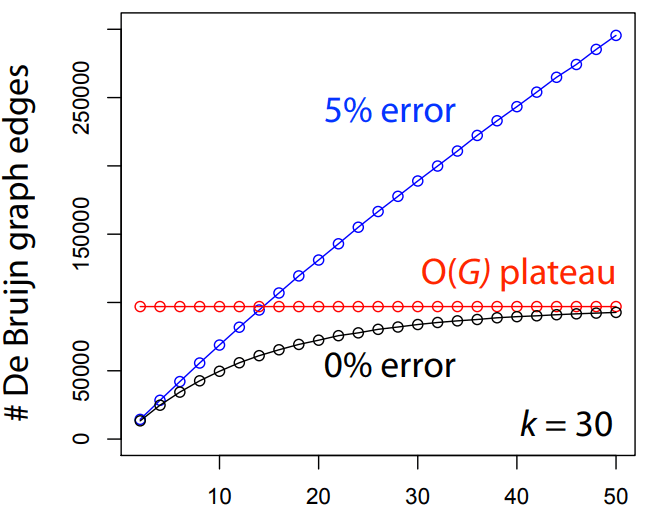
The greedy approach is computationally efficient but fails for large, complex genomes.
Full pairwise comparisons between reads require operations
where nnn is the number of reads
As our number of reads increases, our time to find overlaps dramatically increases
However, the number of reads also improves our assembly
By decomposing reads into k-mers, we can:
Instead of comparing whole sequences, we can compare k-mers!
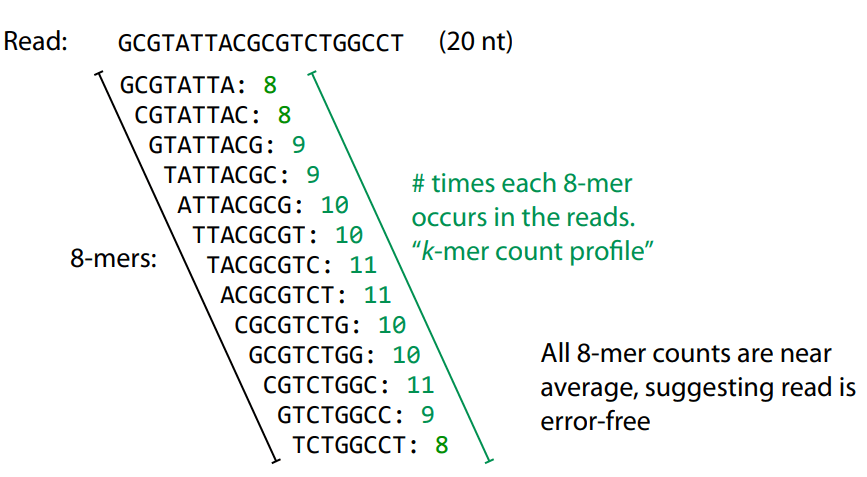
A k-mer is a substring of length kkk extracted from a sequence
Example: For the sequence ATCGT, the 3-mers are ATC, TCG, CGT.
GGCGATTCATCG
Spectrum with k = 3
GGC
GCG
CGA
TCG
ATC
GAT
ATT
TTC
TCA
CAT
All 3-mers
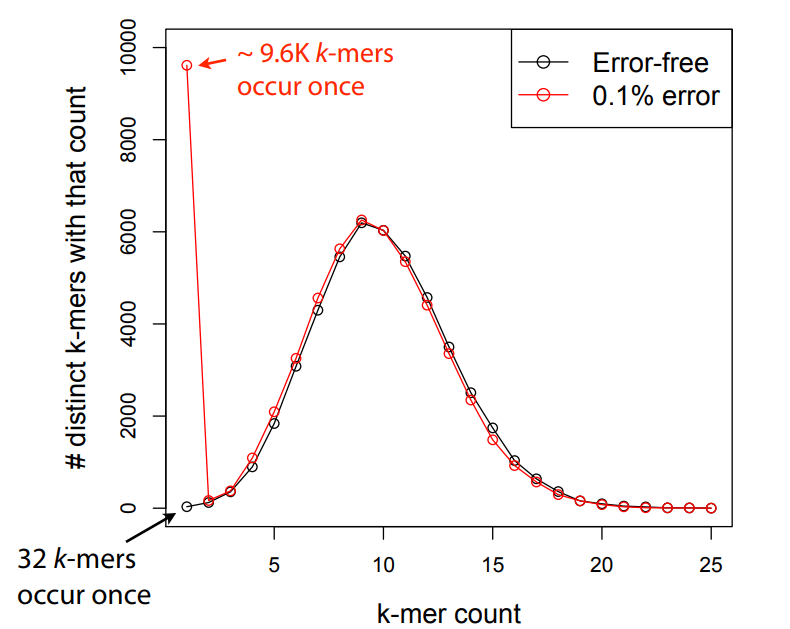
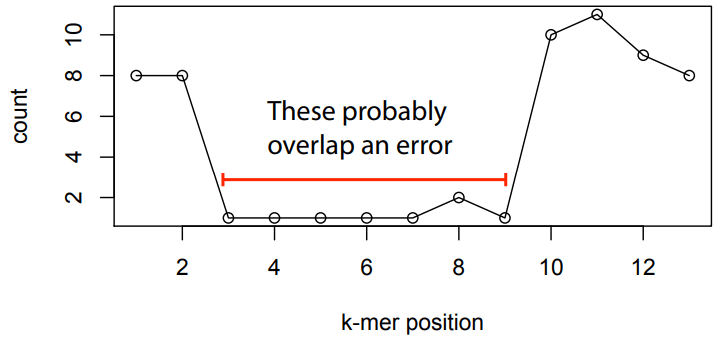
Sequencing errors affect only a few k-mers in a read, not the entire sequence.
Longer k-mers provide specificity, while shorter k-mers ensure sensitivity.
Even if a single read has errors, most k-mers will match correctly to others.
Building graphs
Node
Represents a single entity
Edge
Represents a connection (possibly with a direction)
"tomorrow and tomorrow and tomorrow"
Let's build a directed multigraph:
K-mer is a substring of length k
(We will cheat here and write down just unique words)
tomorrow
and
AATGGCGTA
AAT
ATG
GGC
GCG
TGG
CGT
GTA
AATG
ATGG
TGGG
GGCG
GCGT
CGTA
5'
3'
Step 1:
Let's use kedge = 4
Step 2:
AATG
L
R
Step 3: Repeat
ATGG
TGGG
GGCG
GCGT
CGTA
Take left and right k-1 mer and make two connected nodes (knode = 3)
Build k-mers
CGTAAAT
Build a De Bruijn graph with k = 3
CGT
GTA
TAA
AAA
AAT
CG
GT
TA
AA
AT
5' AATGGCGTA 3'
5' CGTAAAT 3'
Read 1
Read 2
Let's use nodes of length 4
AAT
ATG
GGC
GCG
TGG
CGT
GTA
TAA
AAA
Note: This is a circular genome
Frist, build the De Bruijn graph for
Read 1
Add edges and any new k-mers from
Read 2
5' AATGGCGTA 3'
5' CGTAAAG 3'
5' TAAAGGCGAA3'
Read 1
Read 2
Read 3
AAT
ATG
GGC
GCG
TGG
CGT
GTA
TAA
AAA
AAG
CGA
GAA
AGG
5' AATGGCGTA 3'
5' CGTAAAG 3'
5' TAAAGGCGAA3'
Read 1
Read 2
Read 3
AAT
ATG
GGC
GCG
TGG
CGT
GTA
TAA
AAA
AAG
CGA
GAA
AGG
2
2
2
1
1
1
1
1
2
1
1
1
GATTAC
TACAGATT
AGATTAC
TACCGG
GGATTA
The solution is on the next slide (no peeking!)
De Bruijn graphs is one of the most missed questions on assessments, let's get some practice
GATTAC
TACAGATT
AGATTAC
TACCGG
GGATTA
TACC
ACCG
CCGG
GATT
ATTA
TTAC
TACA
ACAG
CAGA
AGAT
GGAT
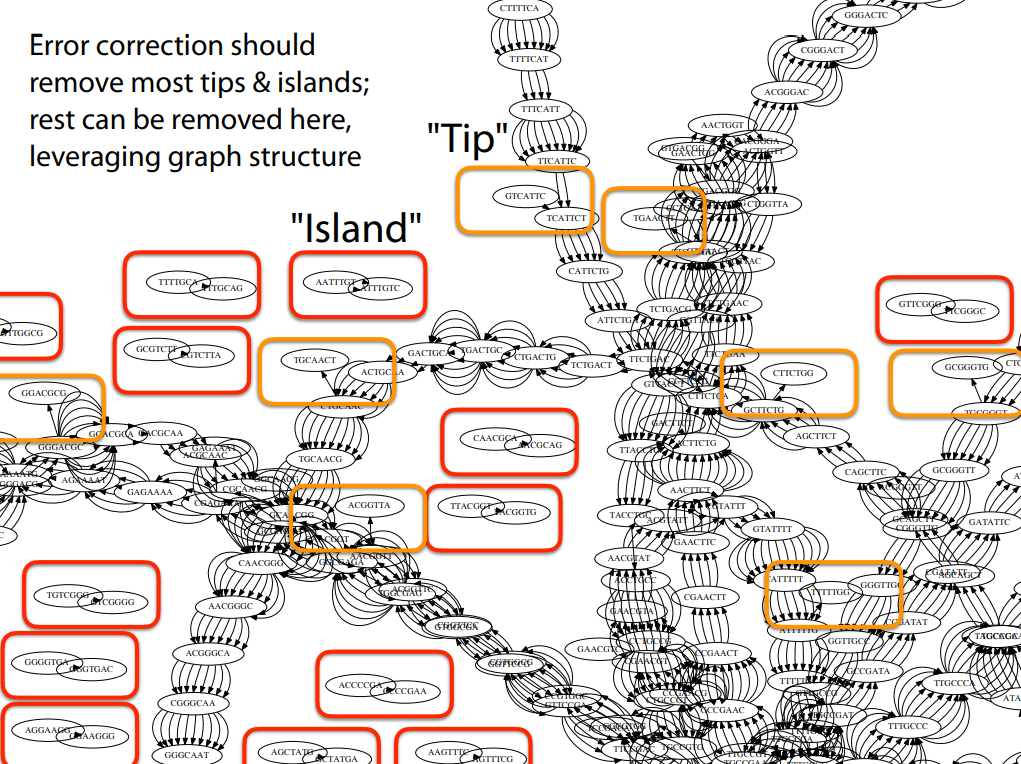
Characteristics
Tips are good starting points of contigs if they have high coverage
Islands are reads we couldn't merge
Traversal is the process of finding contigs (continuous DNA sequences) by walking through the De Bruijn graph
TACC
ACCG
CCGG
GATT
ATTA
TTAC
TACA
ACAG
CAGA
AGAT
GGAT
Edges: Represent k-mer overlaps between nodes.
Nodes: Represent k-mers derived from sequencing reads.
Standard traversal methods, such as breadth-first search (BFS) and depth-first search (DFS), are building blocks for more advanced assembly techniques.
Imagine exploring a maze with this strategy:
A
/ \
B C
/ / \
D E FDFS Traversal from A (one possible order):
Imagine you're dropping a pebble in a pond:
A
/ \
B C
/ / \
D E FDFS Traversal from A (one possible order):
Specialized traversal methods, like Eulerian and Hamiltonian paths, address these challenges.
Lecture 04A:
Genome annotation -
Foundations
Lecture 03B:
Genome assembly -
Methodology
Today
Tuesday
Quiz 01
AAT → ATG → TGG → GGC forms a single path when traversing overlaps.AATGGCGT.This is a valid superstring, but why would we want the shortest?
BAAAABBBAABAABBBBBAAABAB
Talk with your neighbors
Overlap maximization
Repeat resolution
Resolves repeats by favoring collapsed arrangements
Evolutionary pressure
Most genomes have selective pressure to be efficient
Suppose we have a collection of strings (i.e., reads)
(In CS, we call a sequence of characters a string)
BAA
AAB
BBA
ABA
ABB
BBB
AAA
BAB
We want to assemble these strings into a single, continuous string (i.e., contig)
What's the easiest way?
Concatenate
BAAAABBBAABAABBBBBAAABAB
Done!
Well, no.
Right?
This is called a "superstring"
Error correction