BIOSC 1540: L04B (Gene prediction)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Jan 30, 2025
Lecture 04B
Gene prediction
Methodology
Assignments
Quizzes
CBytes
ATP until the next reward: 1,783
Gene prediction is essential for genome annotation and understanding gene function.
Predicted genes
DNA sequences contain a mix of coding and noncoding regions.
However, reliance on fixed patterns limited accuracy in complex genomes
In the early days (1980s), gene prediction was based on hardcoded rules
Fickett, J. W. (1982). Recognition of protein coding regions in DNA sequences. Nucleic acids research, 10(17), 5303-5318.
For example, they would search for
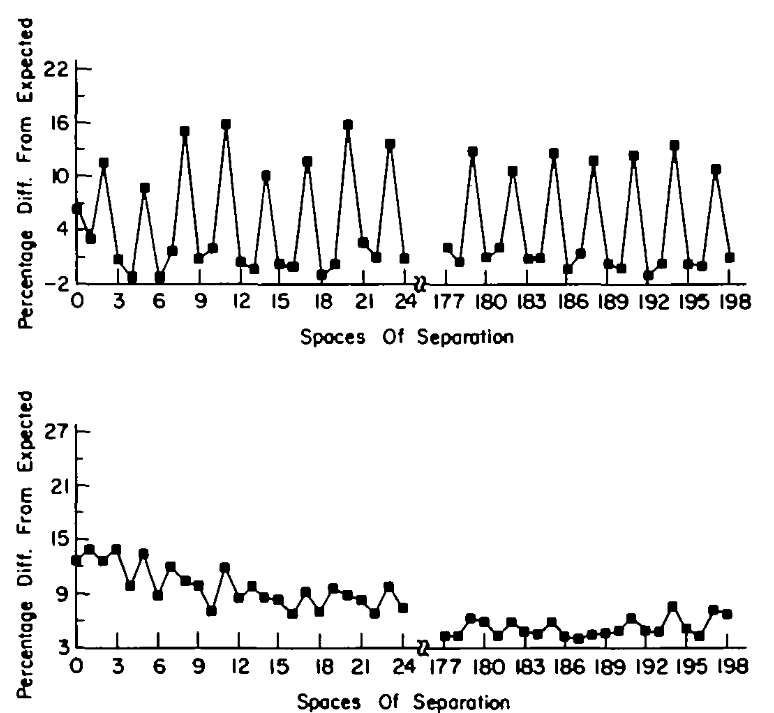
Autocorrelation of T in sequences
Coding
Non-coding
Many non-coding sequences contain patterns that mimic coding sequences, leading to high false positive rates
Not all genes follow the same start/stop codon rules, and promoter motifs are not always well-defined.
Some genes overlap or exist within other genes, making simple start/stop rules unreliable.
Conditional probability
Instead of using fixed rules, we use probabilistic models that quantify uncertainty
These models assign probabilities based on multiple features:
Gene prediction inherently relies on dependencies between nucleotides, codons, and genomic regions
Independent Events: The probability of one event does not affect another
Example: Rolling a die twice—each roll is unaffected by the previous one
Dependent Events: The probability of one event depends on another
Example: A sequence with a high GC-ratio is more likely to belong to a coding region.
Genes often have higher GC content that surrounding non-coding regions
However, not all GC-rich regions are genes, and not all genes are GC-rich
Our goal is to update our belief (i.e., probability) about "gene-ness" based on a region's GC content
Probability of being GC-rich
Probability that a region is a gene given that it's GC-rich
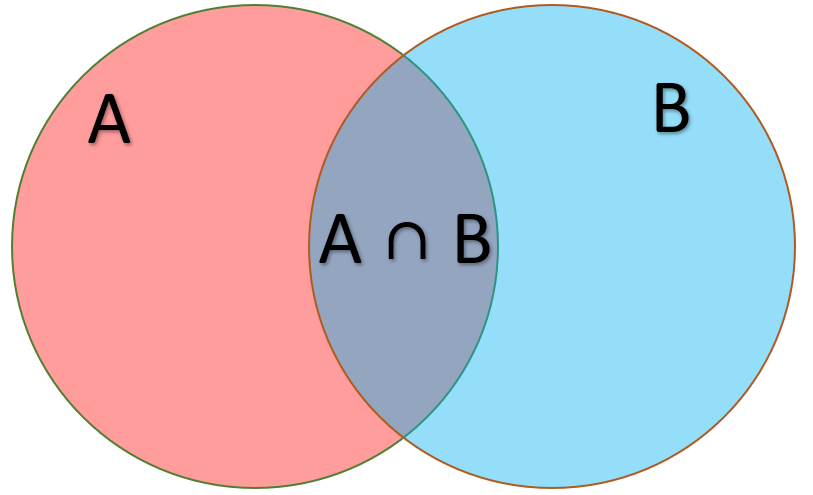
Suppose I want to compute the probability of a region being both GC-rich and a gene
means "and" in set notation
This is the (conditional) probability we want to know
Gene
GC-rich
Gene
GC-rich
Rearranging our equation allows us to compute the probability that a given region could be gene if it's GC-rich
If we have the following information available:
Probability of a random region being a gene and GC-rich
Probability of a random region being GC-rich
We can compute these properties with known data
Why multiple signals?
Objective:
Bayes' theorem
Conditional probability for N signals,
For each new signal, you must compute a new, higher-dimensional intersection over the whole genome.
Conditional probability for one signal,
Data Explosion:
Changing Thresholds: If you redefine “GC-rich” from 60% to 65%, you have to recompute those entire intersections.
Interpretation Issues: Knowing the overlap doesn’t explain how each feature individually shifts the probability that we have a gene.
Conditional probability for N signals (and after some math)
Measure
just once and separately measure
Adding a new signal
just requires
Each
shows how strongly feature
indicates a gene
Advantages
When you want to rank or compare classes based on posterior probability, you can ignore the denominator
Bayes’ Theorem allows us to integrate multiple independent signals
(e.g., GC-richness, codon bias) to update the probability that a region is a gene
However, DNA sequences also have a "sequential" aspect to them (e.g., promoters, ribosomal binding sites, etc.)
While effective for multiple independent features, the Bayesian approach doesn't account for the contextual dependencies between consecutive nucleotides or regions
Markov Models provide a framework to incorporate these sequential dependencies, allowing for more accurate and context-aware gene prediction
What is a sequential dependency?
Markov models provide a way to quantify and predict these sequence patterns.
Examples in Biology:
A Markov Model is a stochastic model that describes a sequence of possible events where the probability of each event depends only on the state attained in the previous event.
Real-World Example:
Components of a Markov Chain:
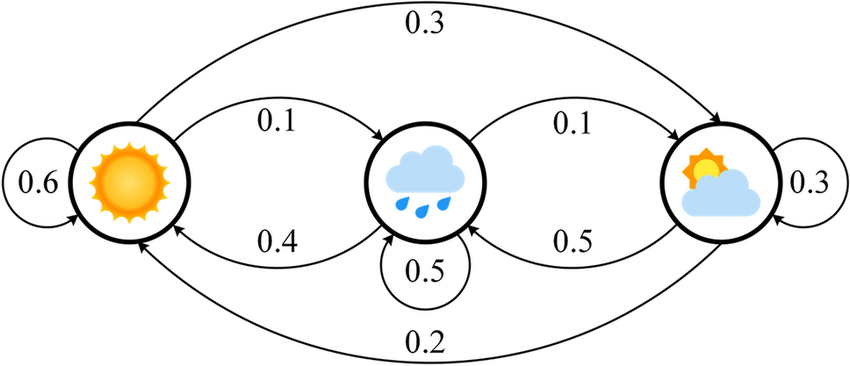
Visual Representation:
Interpretations:
If it's sunny today, it's 60% likely tomorrow will be sunny
If it's cloudy today, it's 50% likely tomorrow will be rainy
First order
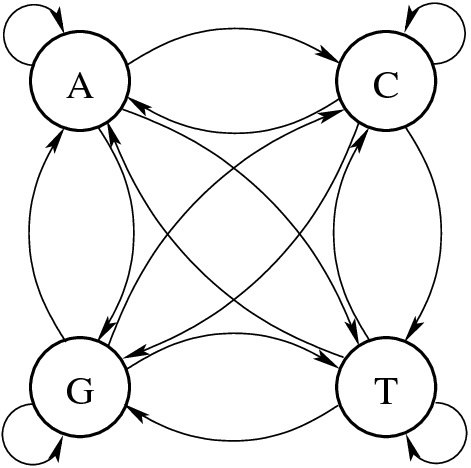
Transition Probabilities: P(A∣G)P(A | G)P(A∣G), P(T∣C)P(T | C)P(T∣C), etc.
The likelihood of observing a nucleotide should depend on the preceding nucleotide
States: {A, C, G, T} – The four nucleotides.
0.3
0.2
0.3
0.2
0.2
0.3
0.1
0.4
0.4
0.1
0.4
0.1
0.1
0.3
0.2
0.4
| A | C | G | T | |
|---|---|---|---|---|
| A | 0.3 | 0.2 | 0.3 | 0.2 |
| C | 0.2 | 0.3 | 0.1 | 0.4 |
| G | 0.4 | 0.1 | 0.4 | 0.1 |
| T | 0.1 | 0.3 | 0.2 | 0.4 |
Instead of a graph, we can represent this as a transition matrix
Interpretation:
Current
Next
Genes (i.e., coding regions) generally have high GC content due to codon biases
Thus, we could assume that coding regions have higher P(G∣C) and P(C∣G)P(C | G)P(C∣G)
Non-coding regions would then have more random nucleotide distributions with less GC bias
| A | C | G | T | |
|---|---|---|---|---|
| A | 0.2 | 0.3 | 0.4 | 0.1 |
| C | 0.1 | 0.4 | 0.3 | 0.2 |
| G | 0.1 | 0.4 | 0.4 | 0.1 |
| T | 0.1 | 0.3 | 0.4 | 0.2 |
Current
Next
| A | C | G | T | |
|---|---|---|---|---|
| A | 0.3 | 0.2 | 0.3 | 0.2 |
| C | 0.2 | 0.3 | 0.1 | 0.4 |
| G | 0.4 | 0.1 | 0.4 | 0.1 |
| T | 0.1 | 0.3 | 0.2 | 0.4 |
Current
Next
Step 1: Train two Markov models—one for coding DNA and one for non-coding DNA.
Step 2: Compute the probability of the observed sequence (S) if it's
Coding (C)
Non-coding (N)
Step 3: Assign the sequence to the model with the higher likelihood
versus
or
If this sequence follows a C or N pattern, the probability will be higher
Limited Context Awareness: FOMMs consider only the immediately preceding nucleotide, preventing them from capturing the inherent triplet codon structure of protein-coding sequences.
Frame-Shift Misclassification: Sequences that deviate from typical single-nucleotide transitions, such as those affected by insertions or deletions, may be incorrectly classified, leading to misidentification of coding regions.
Randomized Nucleotide Transitions: Since transitions occur between individual bases rather than codons, FOMMs do not distinguish between meaningful codon sequences and arbitrary base order.
Higher order
A k-th order Markov model considers k previous nucleotides when predicting the next k
A third-order Markov Model is codon-based
| ATG | CCT | GTA | TTA | |
|---|---|---|---|---|
| ATG | 0.2 | 0.3 | 0.4 | 0.1 |
| CCT | 0.1 | 0.4 | 0.3 | 0.2 |
| GTA | 0.1 | 0.4 | 0.4 | 0.1 |
| TTA | 0.1 | 0.3 | 0.4 | 0.2 |
Current
Next
Transition probabilities reflect valid codon structures, ensuring a more biologically accurate model
Models capture statistical biases inherent in real genes, such as the rarity of stop codons within coding regions
(Not all of them)
We can bake in the idea that the region (i.e., coding or non-coding) influences codon transitions directly into one model
Coding (C)
Non-coding (N)
or
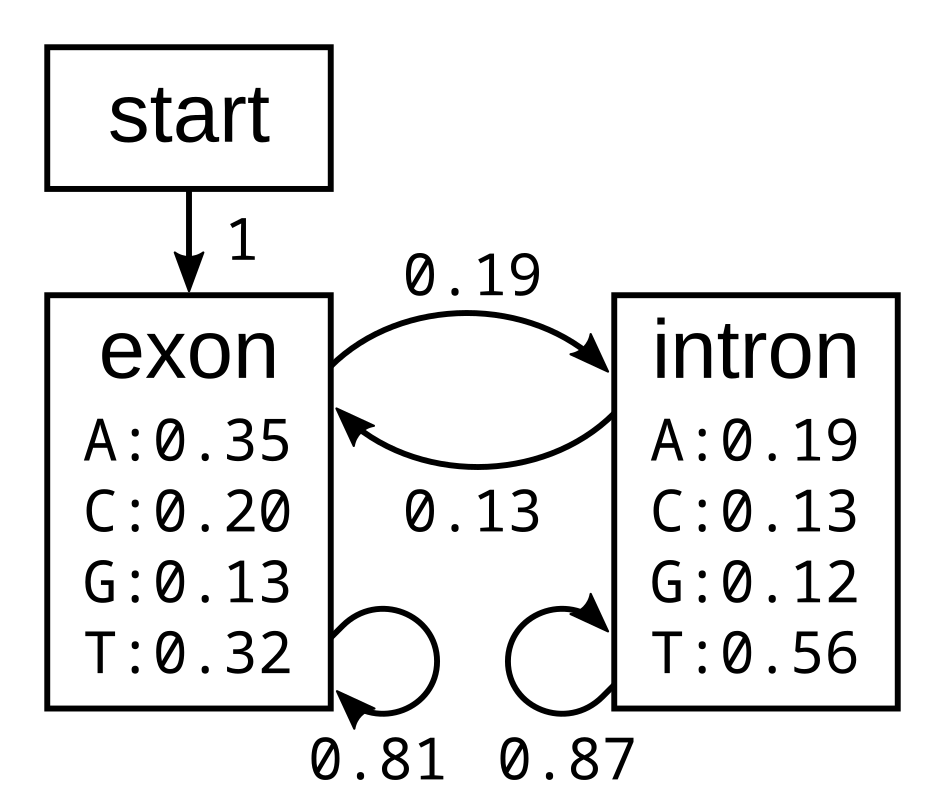
Hidden States are biological states (e.g., coding vs. noncoding DNA) we are trying to determine
What we can still observe are k-mer transition probabilities in our genome
By directly including hidden states in our model, we can directly infer the optimal sequence of hidden states to explain our observed state transitions
States
Transition Probabilities
Emission Probabilities
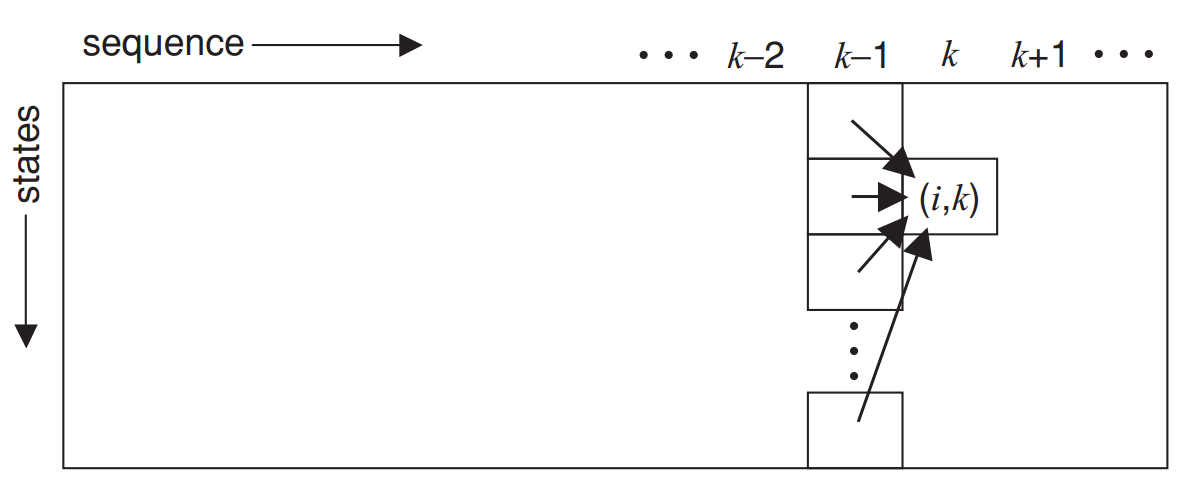
Instead of computing all possible paths, Viterbi keeps track of the best path so far.
The Viterbi algorithm finds the best possible sequence of hidden states that explains the observed sequence
It is essential for determining which nucleotides belong to a gene
This is called dynamic programming, and we will cover this topic next week!
Step 1: Define Components
Step 2: Initialize Probabilities
Step 4: Traceback
Step 3: Fill in the Table
Lecture 05A:
Sequence alignment -
Foundations
Lecture 04B:
Gene prediction -
Methodology
Today
Tuesday