BIOSC 1540: L05A (Alignment)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Feb 4, 2025
Lecture 05A
Sequence Alignment
Foundations
Assignments
Quizzes
CBytes
ATP until the next reward: 1,653
Homology
Homologous sequences share a common ancestor, even if they have diverged over time.
Homology helps transfer knowledge from well-studied genes to newly discovered ones.
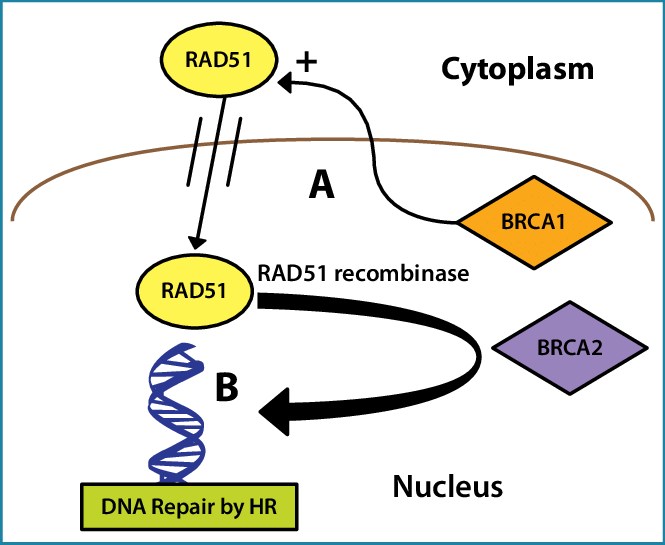
Example: The identification of BRCA1 as a breast cancer gene was based on identifying its association with RAD51, which function was known due to its high homology with yeast DNA repair
They typically perform the same function across species but may accumulate minor adaptations.
Orthologs arise from speciation events, meaning a single ancestral gene diverges into different species.
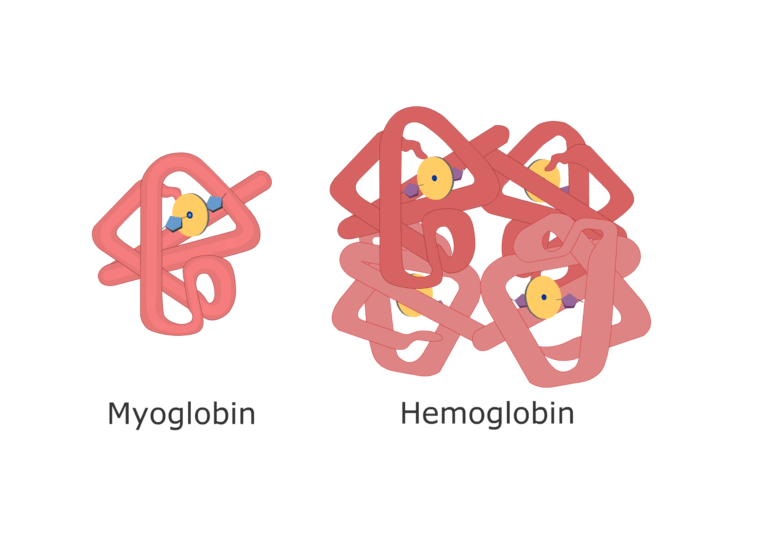
Example: The hemoglobin gene in humans and mice is orthologous, both encoding oxygen-carrying proteins in red blood cells.
Paralogs originate from duplication events, which allows one copy to retain the original function while the other copy can
Paralogs drive gene family expansions, leading to specialized and diverse biological functions.
Examples of paralog-driven functional diversification:
Homology applications
Conserved motifs suggests similar functional roles in different organisms.
For example, if a newly discovered protein aligns with a known enzyme, it likely shares the same biochemical function.
Homology-based searches (e.g., BLAST) rapidly annotate unknown sequences by comparing them to well-characterized databases.
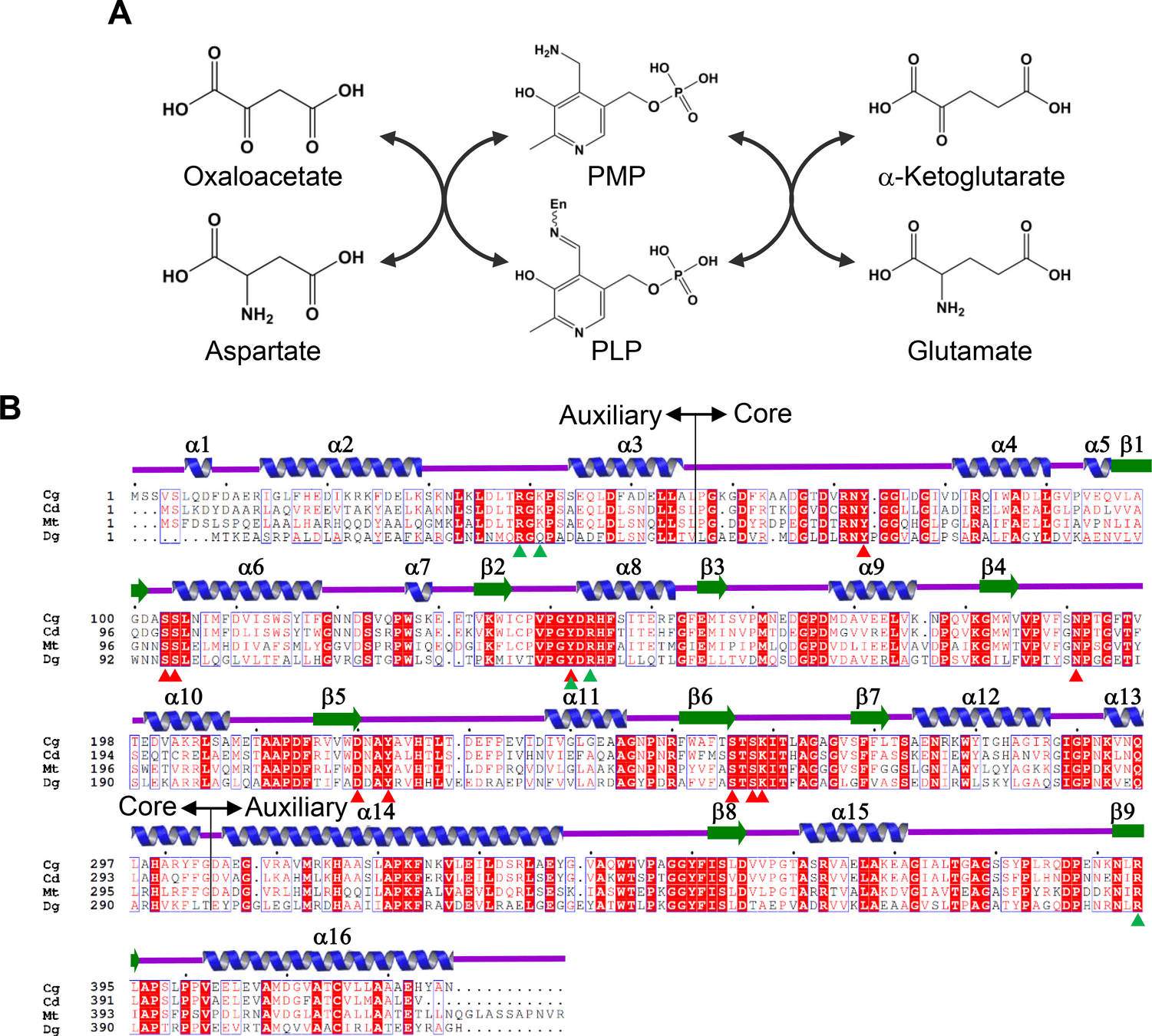
Aspartate aminotransferase (AspAT)
Conserved residues that bind to PLP cofactor are shown with triangles
Structural homology models unknown proteins based on alignment with known structures.
Highly conserved residues often indicate key structural or catalytic sites.
We will cover this in Lectures 11A and 11B
Protein threading techniques align sequences to structural databases like the Protein Data Bank.
AlphaFold uses sequence alignments as inputs to its deep learning model
Importance in research:
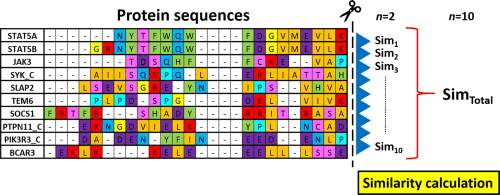
Match and mismatches
Alignment algorithms compare sequences to identify conservation, mutations, and functional domains.
Alignment patterns reflect evolutionary events, including mutations, conservation, and sequence divergence
Highly conserved sequences often correspond to:
Matches (|) indicate strong evolutionary constraints, meaning the sequence is critical for function.
CGACGATTCTATAGTCTAACATGCGAGCGTGACGAATAAAAGATCTCGCG
||||||||||||||||||||||||||||||||||||||||||||||||||
CGACGATTCTATAGTCTAACATGCGAGCGTGACGAATAAAAGATCTCGCGPoint mutations can be neutral, beneficial, or deleterious.
Mismatches occur when different residues are aligned
CGCGATTCTATAGTCTAACATGCGAGCGTGGAAAAAAGATCTCGCG
|| |||||||| | | | | | ||| ||| |||
CGACGATCTATAGTAACATGCGAGCGTGACGAATAAAAGATCTGCGInsertions and deletions
Functional and evolutionary impact:
Causes of insertions:
Gaps are used to indicate insertions
CGACGATTCTATAGTC-------------TGACGAATAAAAGATCTCGCG
|||||||||||||||| |||||||||||||||||||||
CGACGATTCTATAGTCTAACATGCGAGCGTGACGAATAAAAGATCTCGCGCauses of deletions:
Functional and evolutionary consequences:
Gaps are used to indicate deletions
CGACGATTCTATAGTCTAACATGCGAGCGTGACGAATAAAAGATCTCGCG
|||||||||||||||| |||||||||||||||||||||
CGACGATTCTATAGTC-------------TGACGAATAAAAGATCTCGCGIndels play a major role in speciation by modifying gene structure and expression.
Indels can disrupt coding sequences or regulatory elements, leading to disease.
Frameshift mutations caused by small indels result in completely altered protein sequences.
Alignment scores
Alignment algorithms assign numerical scores to quantify how well two sequences align.
Matches receive positive scores (e.g., +1 or +2)
Mismatches receive negative scores (e.g., -1 or -2)
Gaps are heavily penalized (e.g., -2, or -3).
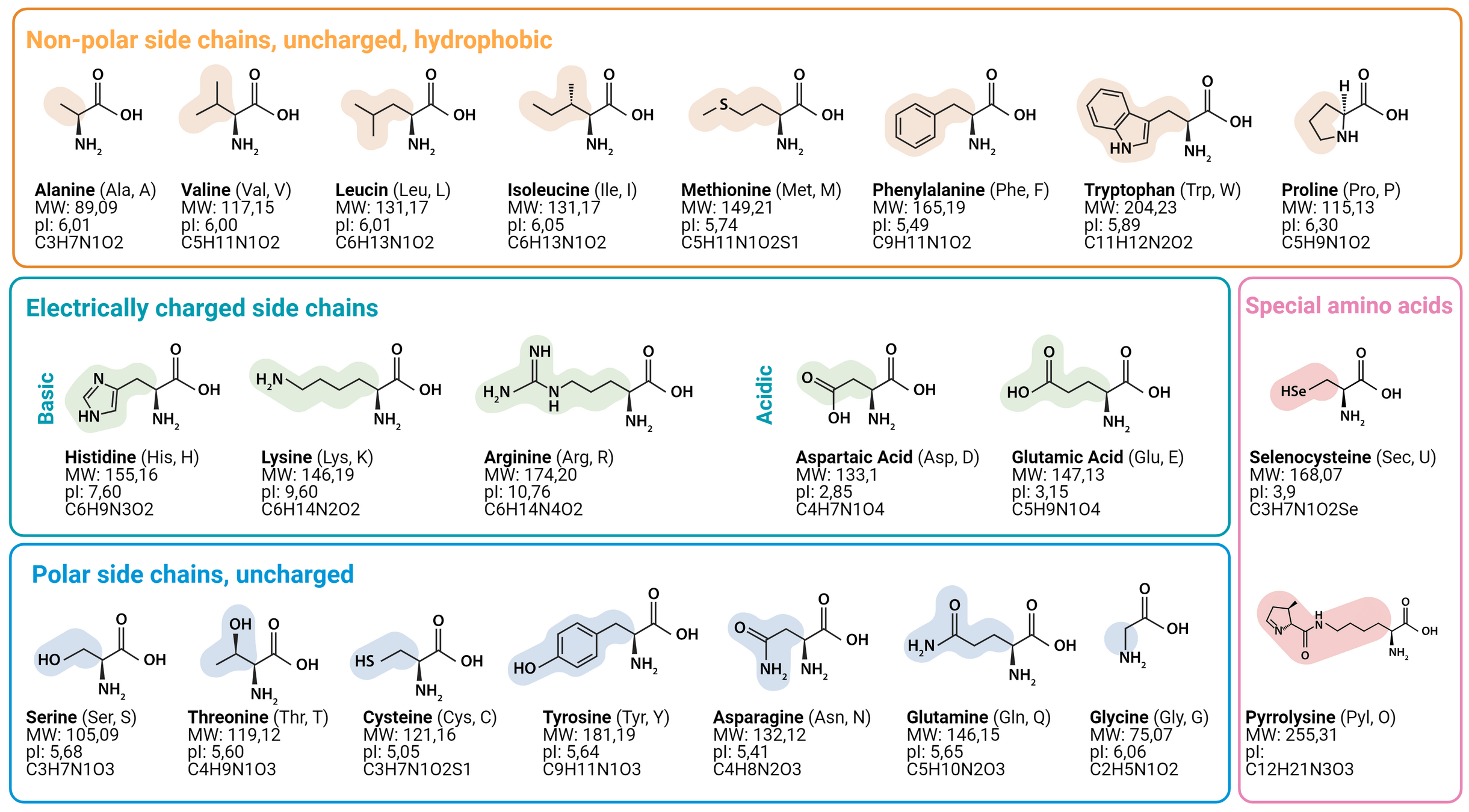
Not all mutations are equally likely—some substitutions occur more frequently due to biochemical properties.
Impact on alignment quality:
Substitution matrices assign different scores to different amino acid replacements based on their evolutionary likelihood.
Two widely used matrices are PAM (Point Accepted Mutation) matrices and BLOSUM (Blocks Substitution Matrix)
Physicochemical properties influence substitution likelihood:
E-values
E-value (Expectation Value): Number of expected random matches in a database search.
Lower E-value = Higher significance (e.g., E = 0.001 means only 1 in 1,000 alignments is due to chance).
Database size affects E-value: Larger databases increase the probability of chance alignments.
Methods like global and local alignment provide different perspectives on sequence similarity.
Pairwise alignment finds the optimal arrangement of two sequences to maximize similarity and minimize differences.
Query 1 ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAA 60
Sbjct 2555705 .....A.....A..AA.T..C..T..C..TAAA...A....C.....G.ACC........ 2555646
Query 61 TTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACT 120
Sbjct 2555645 ...........CT.............A......A.....C..C.GA.C.....GA....A 2555586
Query 121 TTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAAT 180
Sbjct 2555585 C.T.......CA..G..A..T...A.T..T..A..G..G...T.G..A...A.A..T..C 2555526
Query 181 GTTGTACTTACTTC---AGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCT 237
Sbjct 2555525 ..C.....C...AACCA..C.T..--.....C.A.GA....-..A.....T..AA.C... 2555469
Query 238 ATTGAAGATATTTATCAACTACCGGGCCATGTTTTTATATTTGGAGGGCAAACATTATTT 297
Sbjct 2555468 C....T..A...A.AG.GT..T.T..T....................A.....G....AC 2555409
Query 298 GAAGAAATGATTGATAAAGTGGACGACATGTATATTACTGTTATTGAAGGTAAATTTCGT 357
Sbjct 2555408 ....C.........CC.G..A..T..T........C..A..A..A..T..A..G....AA 2555349
Query 358 GGTGATACGTTCTTTCCACCTTATACATTTGAAGACTGGGAAGTTGCCTCTTCAGTTGAA 417
Sbjct 2555348 ..A..C..A...........A..C.....C...A..........C.AA........A... 2555289
Query 418 GGTAAACTAGATGAGAAAAATACAATTCCACATACCTTTCTACATTTAATTCGTAAAAAA 477
Sbjct 2555288 ...C..........A........T..A..G.....A..CT........G.G....G.... 2555229Strengths:
Example: A pairwise comparison of hemoglobin genes between humans and chimpanzees provides insight into species divergence but does not reveal broader evolutionary trends across mammals.
Limitations:
MSA aligns three or more sequences to reveal conserved motifs, functional domains, and evolutionary relationships.
Unlike pairwise alignment, MSA considers multiple substitutions, insertions, and deletions across species.
Example: ClustalW and MUSCLE generate MSAs to compare entire protein families
Sequence alignment identified key conserved residues that are often epitopes
Residues in orange are stem helix epitope region
Lecture 05B:
Sequence alignment -
Methodology
Lecture 05A:
Sequence alignment -
Foundations
Today
Thursday