BIOSC 1540: L04A (Gene prediction)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Jan 28, 2025
Lecture 04A
Gene prediction
Foundations
Assignments
Quizzes
CBytes
ATP until the next reward: 1,783
When you are finished, please hold on to your quiz and feel free to doodle, write anything, tell me a joke, etc. on the last page
In previous lectures, we explored the process of creating contiguous sequences with genome assembly
DNA sequence (i.e., contig)
TACGATCGGATTACGCGTAGGCTAGCTTACGGACTCGATGTACGATCGGATTACG
Gene prediction and genome annotation transform raw sequence data into actionable biological insights, identifying functional elements like genes, regulatory regions, etc.
Predicted genes
Genes encode proteins, enzymes, and non-coding RNAs essential for cellular function
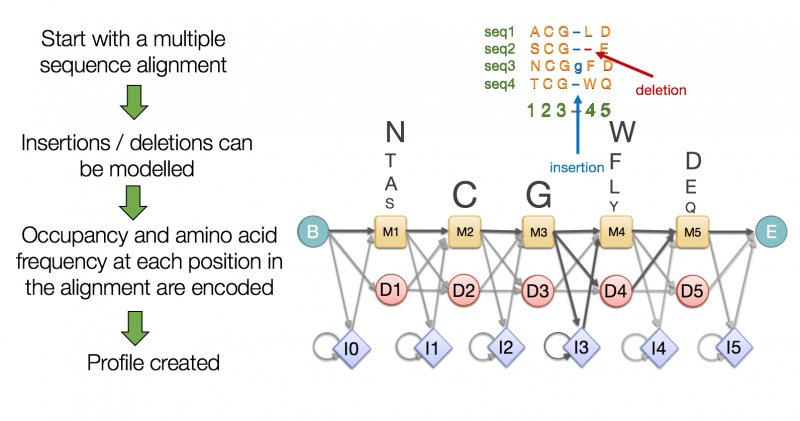
We often use Hidden Markov Models (HMMs) to statistically predict gene locations
(Topic for L04B)
This is also called "structural annotation"
Annotation assigns putative functions to genes through experimental evidence, similarity to known genes, or ab initio predictions.
Functional annotation helps classify genes into pathways (e.g., KEGG), ontologies (e.g., GO terms), and systems (e.g., metabolic networks).
All downstream (i.e., after) analyses are often gene-specific, so any errors here will propogate
Prokaryotes
Most genes are readily identifiable by open reading frame (ORF) detection.
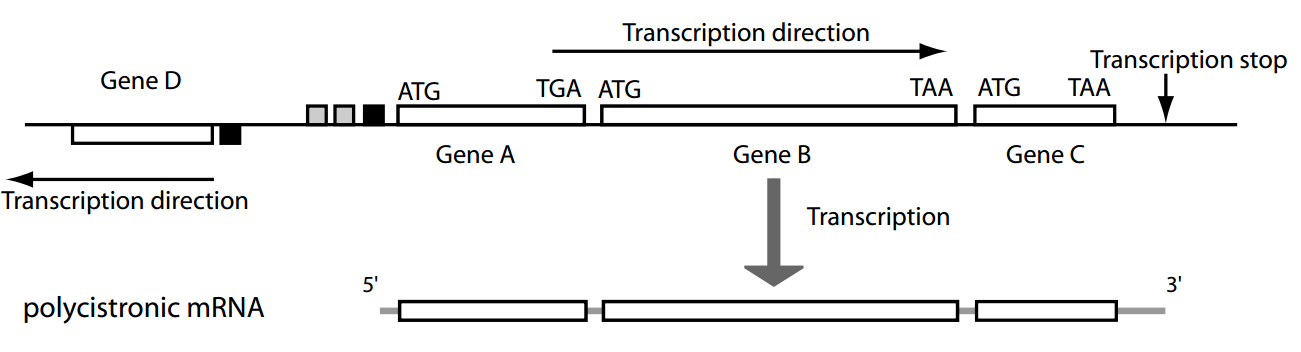
Most genes are organized in operons—clusters of co-transcribed genes under a single promoter.
Promoters
Regulatory sequences
Operon
Polycistronic: coding sequences for two or more polypeptide chains that are transcribed in succession from the same promoter
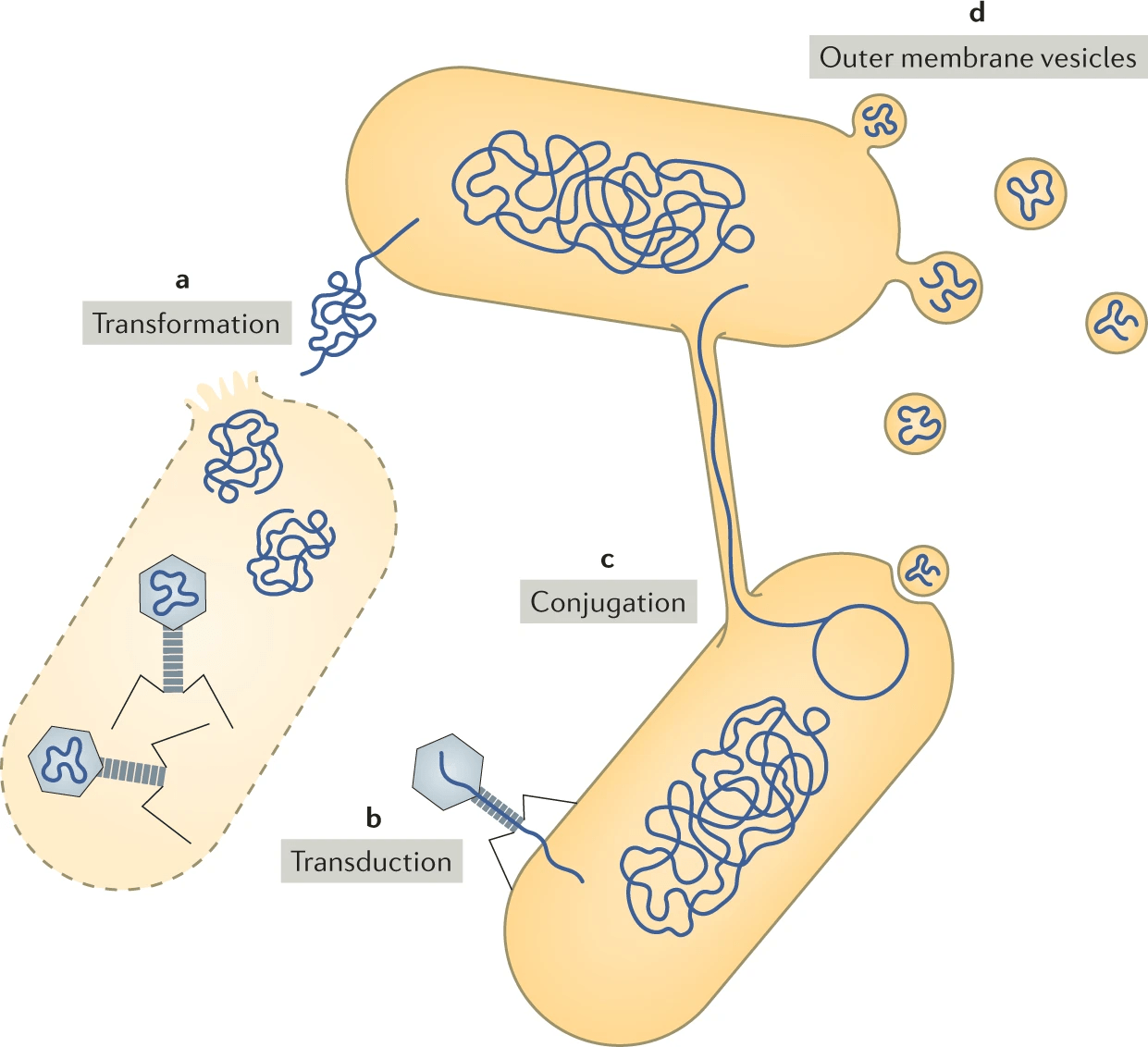
Horizontal gene transfer: Foreign genes may lack organism-specific sequence patterns, complicating detection.
Short genes: Genes shorter than 150 bp are harder to distinguish from random ORFs.
Eukaryotes
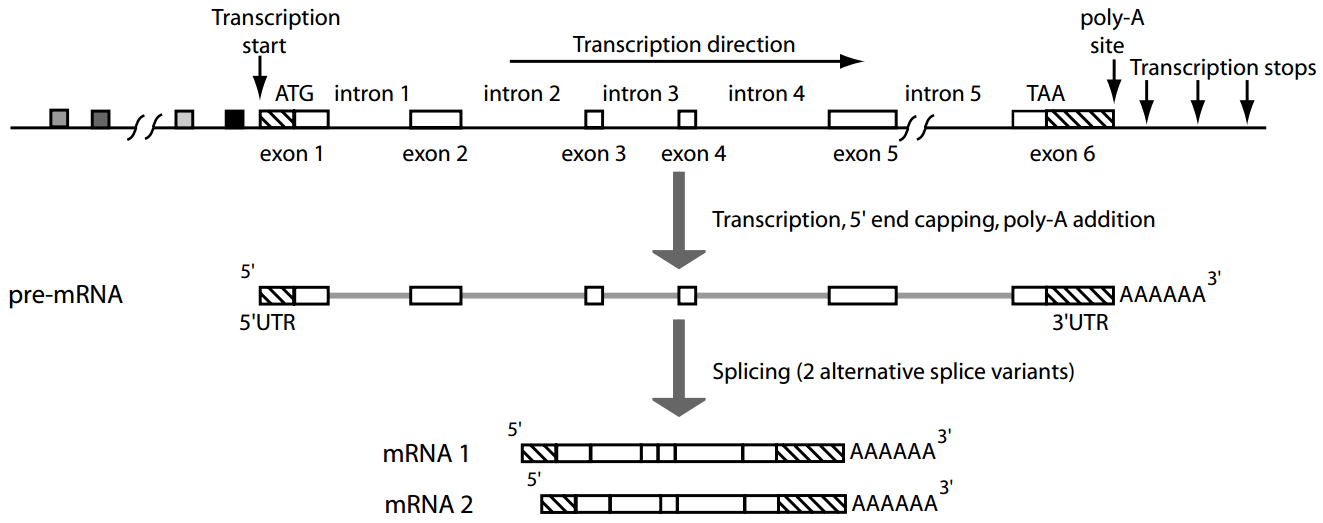
Genes contain introns (non-coding regions) and exons (coding regions), requiring splicing for expression
Intergenic (i.e., between genes) regions are large and often contain regulatory elements (e.g., enhancers, silencers).
Eukaryotic genes undergo splicing which will remove introns and then join exons to form mature mRNA
Gene prediction has to predict intron boundaries, which are often much longer than exons and not always consistent
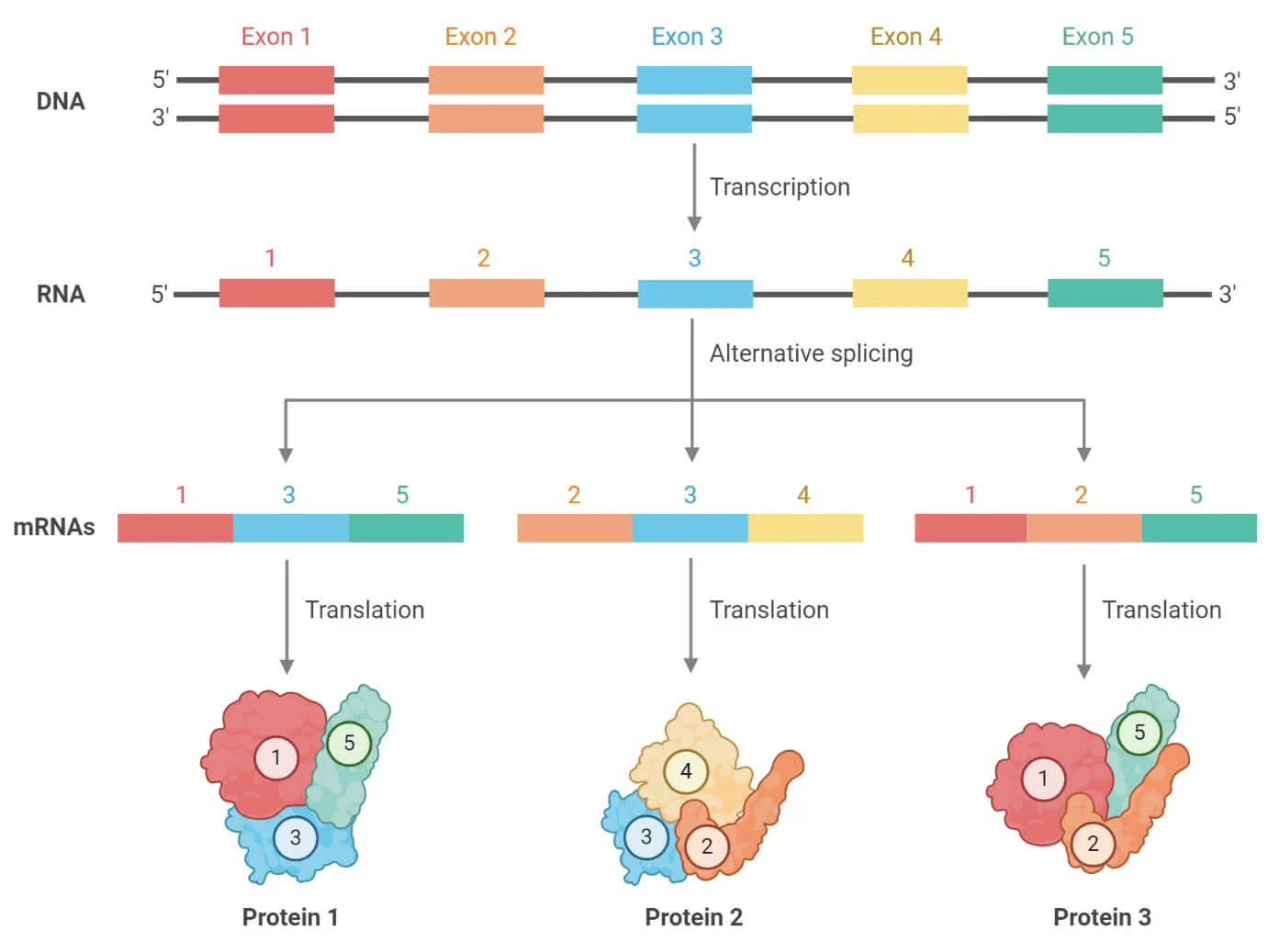
Furthermore, eukaryotes use alternative splicing to join different exons of the same gene to form multiple different proteins
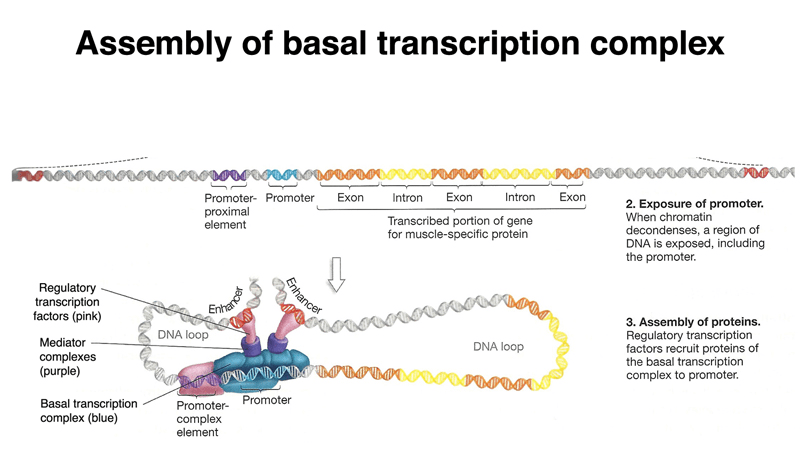
Promoters, enhancers, and silencers regulate transcription but are often far from the gene they control.
These elements lack a universal sequence pattern, making them difficult to identify.
Repetitive sequences: Large portions of eukaryotic genomes are repetitive, often confusing prediction algorithms.
AGCTGATC
TTAGCCGA
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT CGAT
Ab initio
Relies on patterns like:
Detects genes without requiring prior knowledge or reference sequences.
We use HMMs to detect these
Eukaryotes:
Prokaryotes: Compact genomes make ORF detection easier, but short genes and overlapping genes can still pose challenges.
False positives and false negatives are common, especially in large, complex genomes.
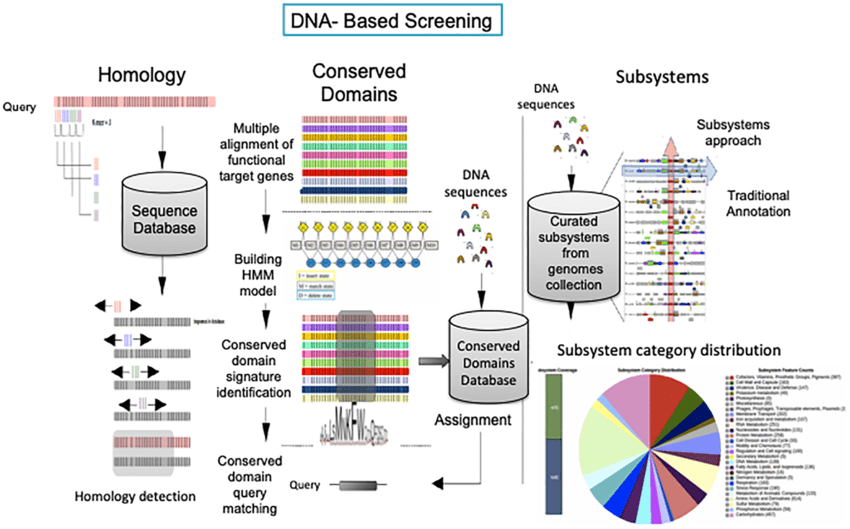
Homology
Tools often use sequence alignment methods (e.g., BLAST, HMMER) to detect homologous genes.
Searches for regions of similarity between the query genome and annotated sequences in databases.
Assumes genes are evolutionarily conserved across species.
Advantages: High accuracy for conserved genes with reliable reference sequences.
Limitations:
Ab initio methods can detect novel genes, filling gaps left by homology-based methods.
Integrated pipelines (e.g., Prokka, AUGUSTUS) use both approaches to produce more reliable results.
Homology-based methods provide functional validation for predictions from ab initio.
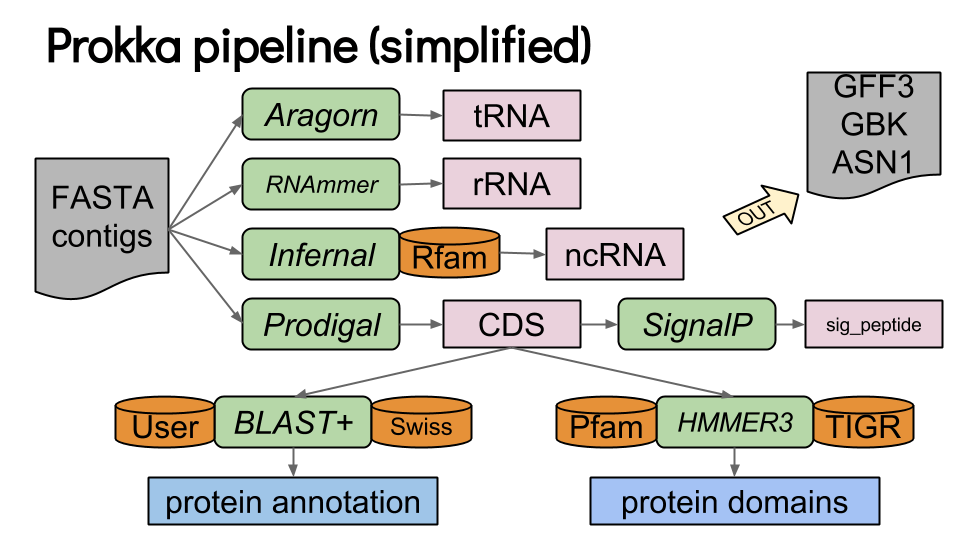
Prokka
Selecting the right tool depends on the organism, genome complexity, and research goals.
Outputs:
Combines ab initio and homology-based methods for prokaryotic genomes.
Annotates coding sequences, tRNAs, rRNAs, and regulatory regions.
Inputs: Assembled genome in FASTA format.
Outputs:
>ECNNONJI_02637 Dihydrofolate reductase
MTLSILVAHDLQRVIGFENQLPWHLPNDLKHVKKLSTGHTLVMGRKTFESIGKPLPNRRN
VVLTSDTSFNVEGVDVIHSIEDIYQLPGHVFIFGGQTLFEEMIDKVDDMYITVIEGKFRG
DTFFPPYTFEDWEVASSVEGKLDEKNTIPHTFLHLIRKKAGUSTUS
Outputs:
Focuses on ab initio gene prediction but integrates hints like RNA-seq data for improved accuracy
Suitable for genomes with limited or no reference annotations
Outputs:
Aligns query sequences to profiles of known genes/proteins in curated databases like Pfam.
Identifies genes based on conserved domains or motifs.
Lecture 04B:
Gene prediction -
Methodology
Lecture 04A:
Gene prediction -
Foundations
Today
Thursday