BIOSC 1540: L08A (Differential gene expression)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Feb 25, 2025
Lecture 08A
Differential gene expression
Foundations
Assignments
Quizzes
CBits
Differential gene expression quantifies changes in gene expression levels between different sample groups or conditions
We have been focused on quantifying gene expression in quantities like Transcripts Per Million (TPM)
Samples
Normal
Cancerous
We could technically directly compare means between our different conditions
However, biological data are inherently noisy, and observed differences may arise by chance
Examples of experimental biases (besides sample variation)
Sequencing depth: Higher depth could appear as higher expression levels simply due to having more data
Batch effects: Processing sampling with different equipment, reagents, times, etc. can show systematic differences
Normal
Cancerous
TPM
Samples
Normal
Cancerous
TPM
Samples
Statistical models can account for variability and separate signal from noise
Hypothesis testing between statistical models provides a quantitative way to compare conditions
After fitting a statistical model, we need to perform hypothesis testing to see if the difference in expression between conditions is statistically significant
Gene expression
Normal
Cancerous
We reject the null hypothesis when our statistical test demonstrates that the observed difference, if any, is unlikely to have happened by random chance
Null Hypothesis (H₀): There is no difference in gene expression between the two conditions
Alternative Hypothesis (H₁): There is a significant difference in gene expression between the conditions
We have two hypotheses:
What is the probability that any difference is either (1) nonexistent or (2) due to random chance
Probability value (p-value):
The higher the p-value, the more our model supports the null hypothesis
The lower the p-value, the more our model supports the alternative hypothesis
Gene expression
Normal
Cancerous
Gene expression
Normal
Cancerous
Binomial distribution
A statistical model describes how data is expected to behave if H₀ is true.
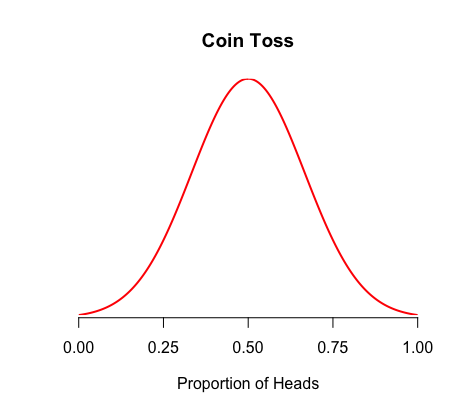
For example, a fair coin flip should result in a normal distribution centered on 50% of each side
This is our statistical model that describes our coin flip observations under H0
We are probably flipping a weighted coin because our observations do not match our H0 statistical model
If we flip a coin 10 million times and our distribution looks like
this
RNA-seq generates count data – the number of RNA fragments that map to each gene
Gene expression
Normal
Cancerous
Example: 573,282 TPM
Discrete data requires us to use special statistical models
What is discrete data?
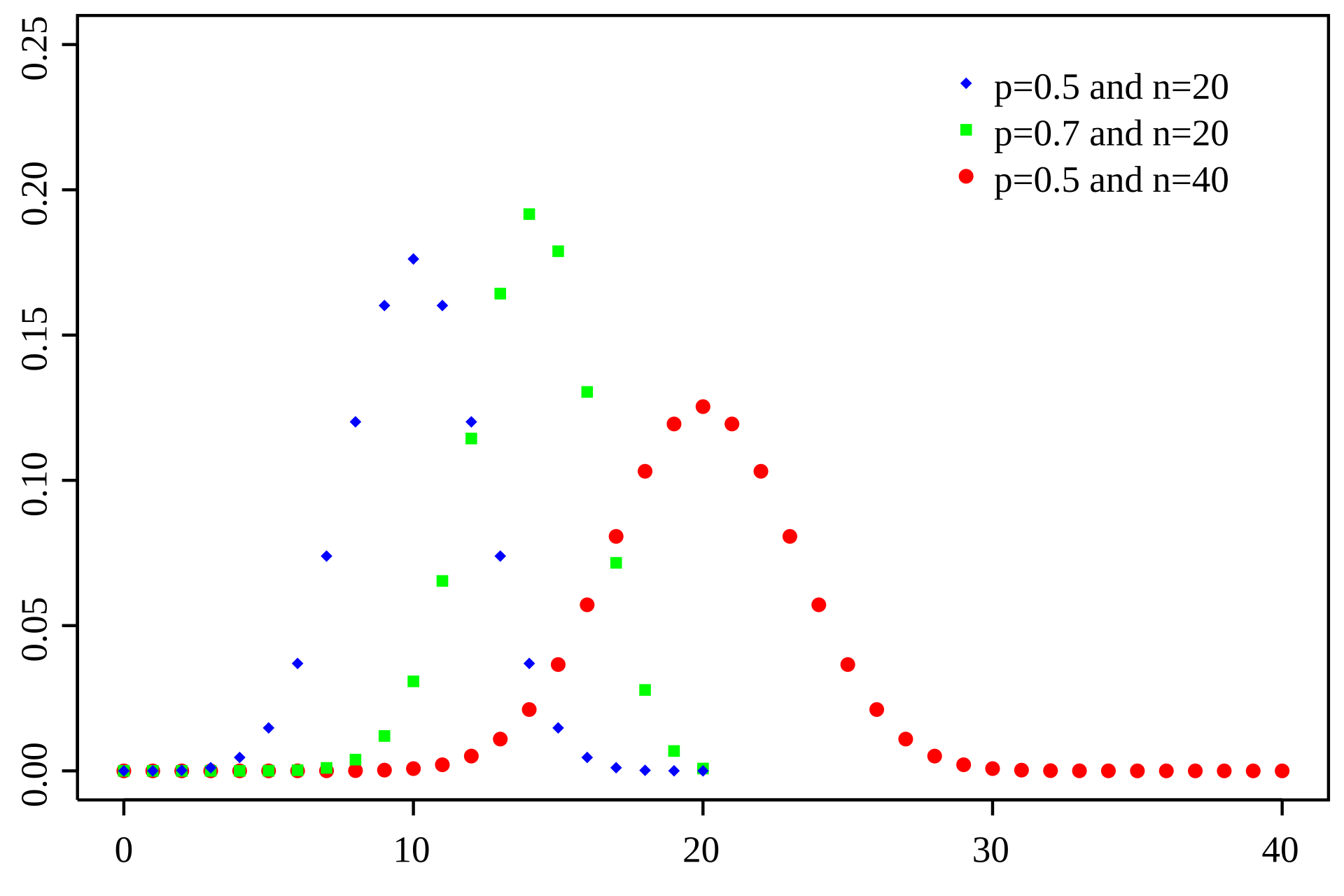
The Binomial distribution models the number of successes in a fixed number of independent trials, where each trial has the same probability of success
Number of trials
Number of successes
Probability
Probability of success
RNA-seq analogy: Each read can be considered a "trial," and the probability that a read maps to a specific gene is the "probability of success."
Poisson distribution
For example, if I have 10 samples from cancerous cells, the binomial distribution assumes they are perfect replicates with no biases
Ignoring sample-to-sample variability can lead to underestimating the true uncertainty in the data
Number of trials
Number of successes
Probability
Probability of success
When sequencing depth is high, n (the total number of reads) becomes very large
Factorials when n is large makes accurate calculations impractical
With very low p, the expected number of successes (reads mapping to a lowly expressed gene) is minuscule compared to n
Calculations with very small probabilities may lead to numerical underflow/imprecise results
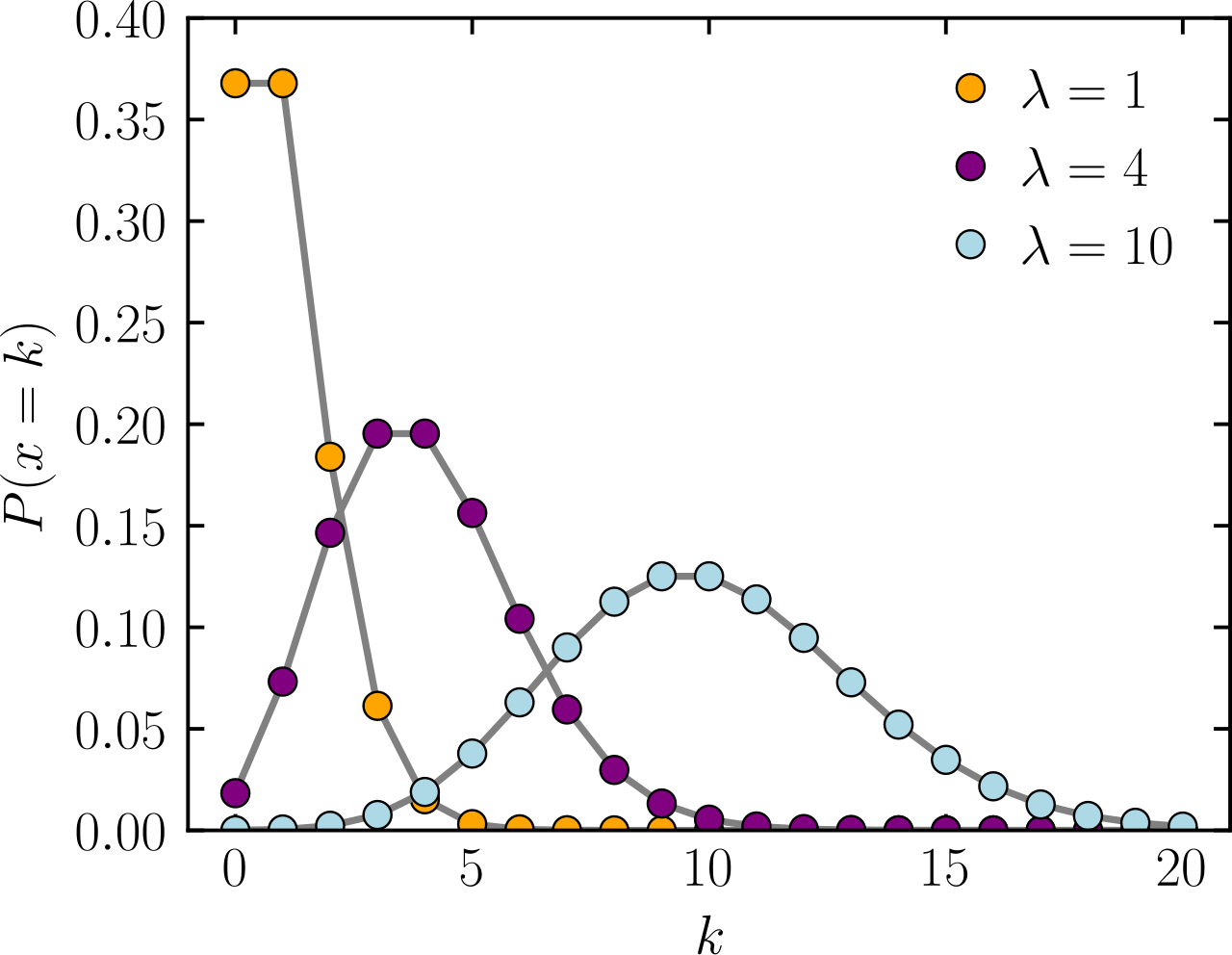
The Poisson distribution is a statistical tool used to model the number of events (i.e., counts) that happen in a fixed period of time or space, where:
Expected average of X
Number of events or counts
Probability
Assuming the constant average rate of success allows some variation around the mean
I.e., sample variation and batch effects
Negative binomial distribution
The expected value (i.e., mean)
When k = 0, the term is zero
Use
You don't need to understand these derivations—just the outcome
When k = 0 or 1, the term is zero
You don't need to understand these derivations—just the outcome
Use
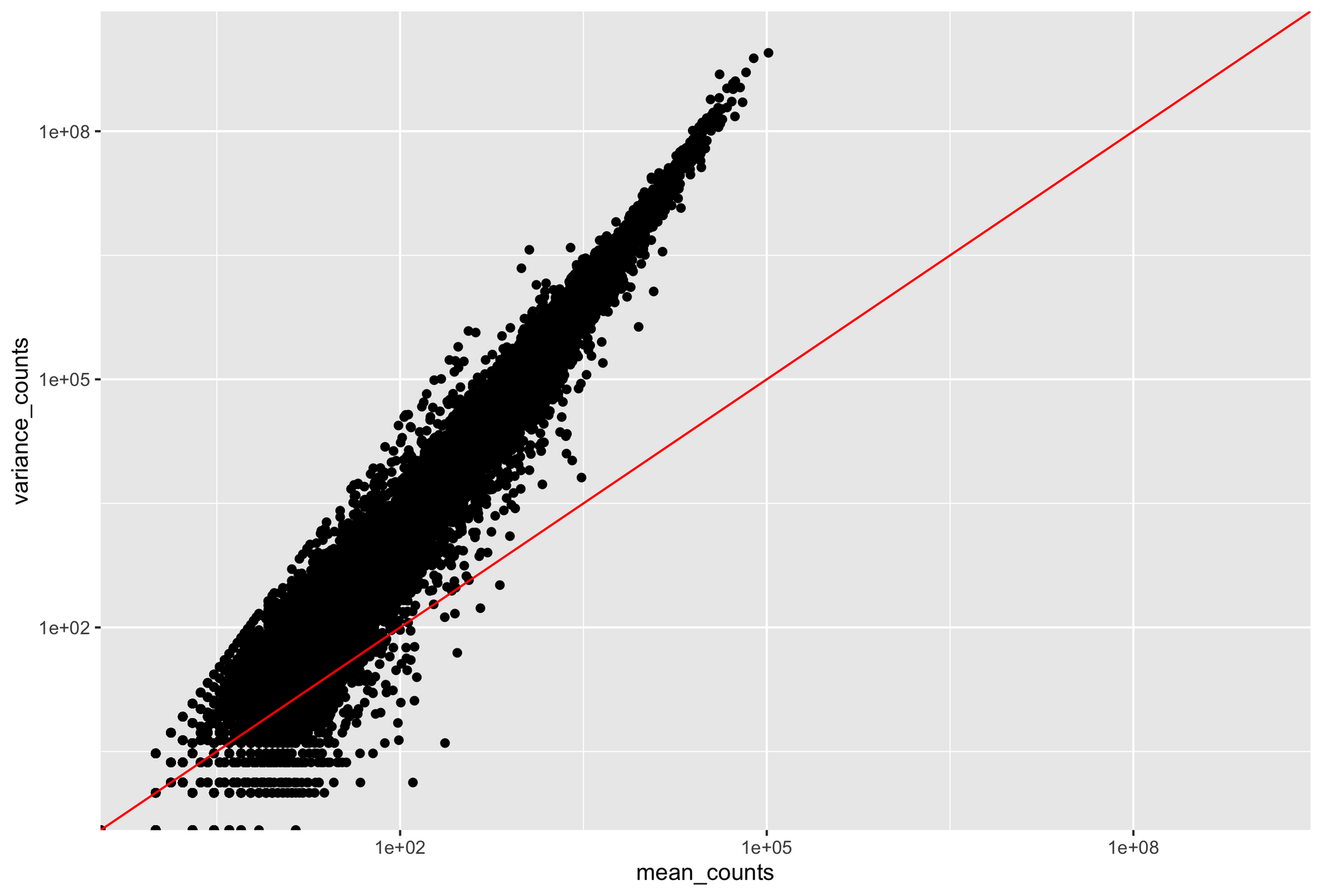
If our variance is different from our mean, our Poisson model breaks down
Higher counts typically have a larger variance
Count mean
Count variance
Mean = variance line
Overdispersion: It happens when the variance in the data is larger than what is predicted by simpler models (e.g., Poisson distribution)
Overdispersion may reflect biological variability between samples not captured by the experimental conditions
Observed number of counts
Mean or expected value of counts
Dispersion parameter, controlling how much the variance exceeds the mean
Gamma function, which generalizes the factorial to floats
If α=0\alpha = 0α=0, the Negative Binomial distribution reduces to the Poisson distribution
RNA-seq data frequently contains zero counts for some genes because not all genes are expressed under all conditions
Most statistical models account for variance, but not that zeros can dominate counts
For example, if we have a high expected mean with Poisson distribution we can still have zeros or very low counts
In these circumstances, we have to use zero-inflated models
We will ignore these for now
A higher product (or joint likelihood) means the model assigns a higher probability to the observed data, indicating a better fit.
The likelihood of model parameters θ\thetaθ given data y\mathbf{y}y is defined as
When individual data points y1,y2,…,yny_1, y_2, \dots, y_ny1,y2,…,yn are independent, the joint probability is calculated by multiplying their individual probabilities:
Multiplying these probabilities aggregates the evidence from each data point, providing a comprehensive measure of how well the model with parameter θ\thetaθ fits all the data
logL(θ)=∑i=1nlogP(yi∣θ)\log L(\theta) = \sum_{i=1}^n \log P(y_i|\theta)
Makes differentiation easier for optimization
Log likelihood
logL(θ)=∑i=1nlogP(yi∣θ)\log L(\theta) = \sum_{i=1}^n \log P(y_i|\theta)
Converts products into sums, reducing computational issues.
At the optimum, the model parameters provide the best explanation of the observed data.
Optimization problem
Bad fit
Good fit
To compute a p-value, a likelihood ratio test (LRT) can be usedβ^1\hat{\beta}_1
The idea is to compare the likelihood of the data under
Log-Likelihood of Negative Binomial
For each condition, you compute the log-likelihoods:
The LRT statistic is:β^1\hat{\beta}_1
The log-likelihood under the null hypothesis (assuming a common mean μ0\mu_0μ0 for both conditions)
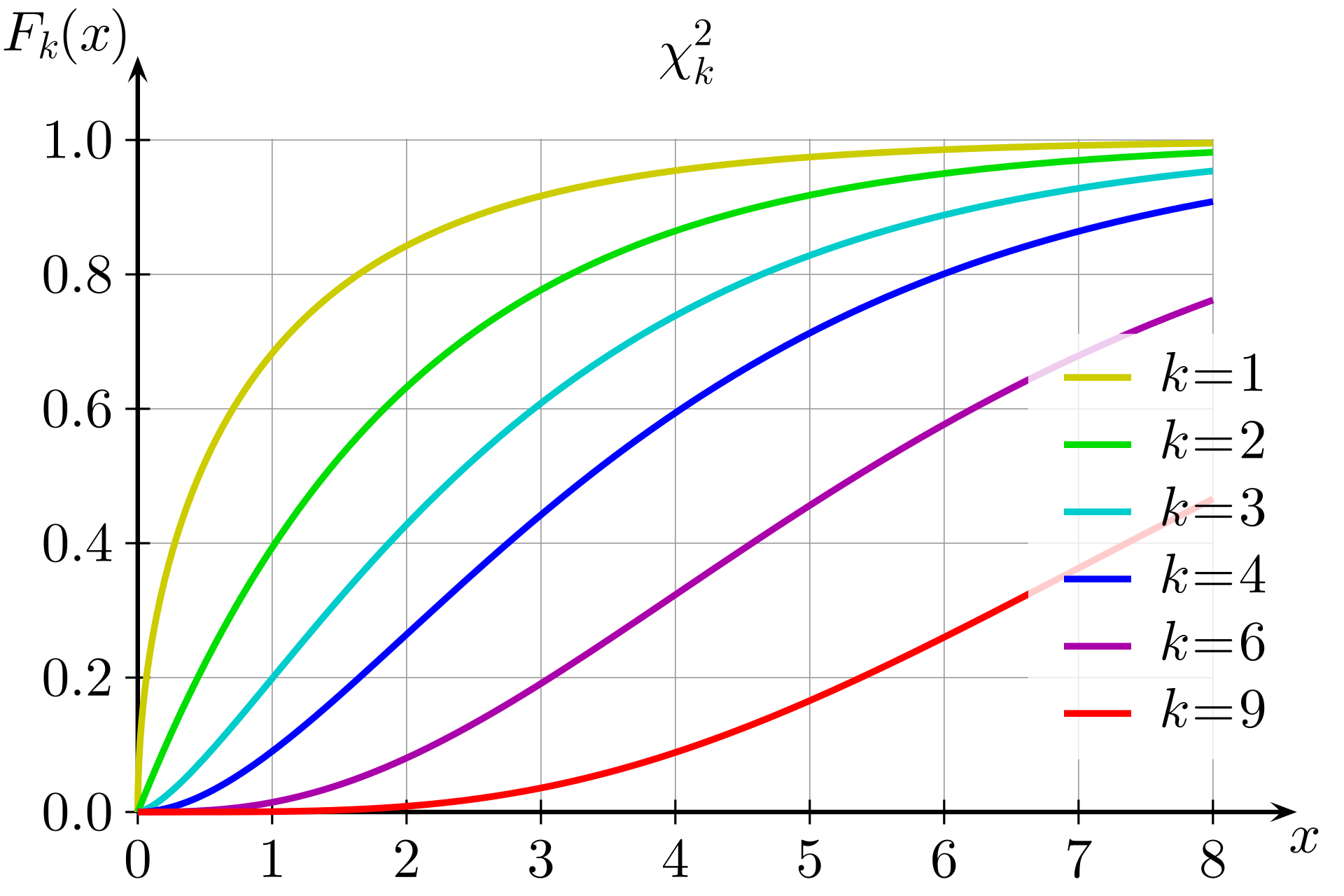
The LRT statistic approximately follows a chi-squared distribution with 1 degree of freedom under the null hypothesis
The p-value is computed as:
k would be 1
Lecture 08B:
Differential gene expression -
Methodology
Lecture 08A:
Differential gene expression -
Foundations
Today
Thursday
Objective: Identify genes differentially expressed between triple-negative breast cancer (TNBC) and hormone receptor-positive breast cancer
Differential gene expression provides statistical tools to identify changes between samples
Findings: TNBC shows upregulation of genes involved in cell proliferation and metastasis.
Implications:
Wald’s Test: A statistical test that helps us determine whether the estimated log fold change between two conditions is significantly different from zero.
Null Hypothesis (H₀): The log fold change between conditions is zero (no difference in expression between the conditions).
Alternative Hypothesis (H₁): The log fold change between conditions is not zero (there is a difference in expression).
For each gene, the Negative Binomial model gives us an estimated log fold changeβ^1\hat{\beta}_1
It also gives us a standard error (SE) for this estimate, which tells us how uncertain we are about the estimate of log fold changeβ^1\hat{\beta}_
The Wald statistic is calculated as
This statistic tells us how many standard deviations the estimated log fold change is away from zero (no difference)