BIOSC 1540: L02A (Sequencing)
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
This is a live streamed presentation. You will automatically follow the presenter and see the slide they're currently on.
Computational Biology
(BIOSC 1540)
Jan 14, 2025
Lecture 02A
DNA sequencing
Foundations
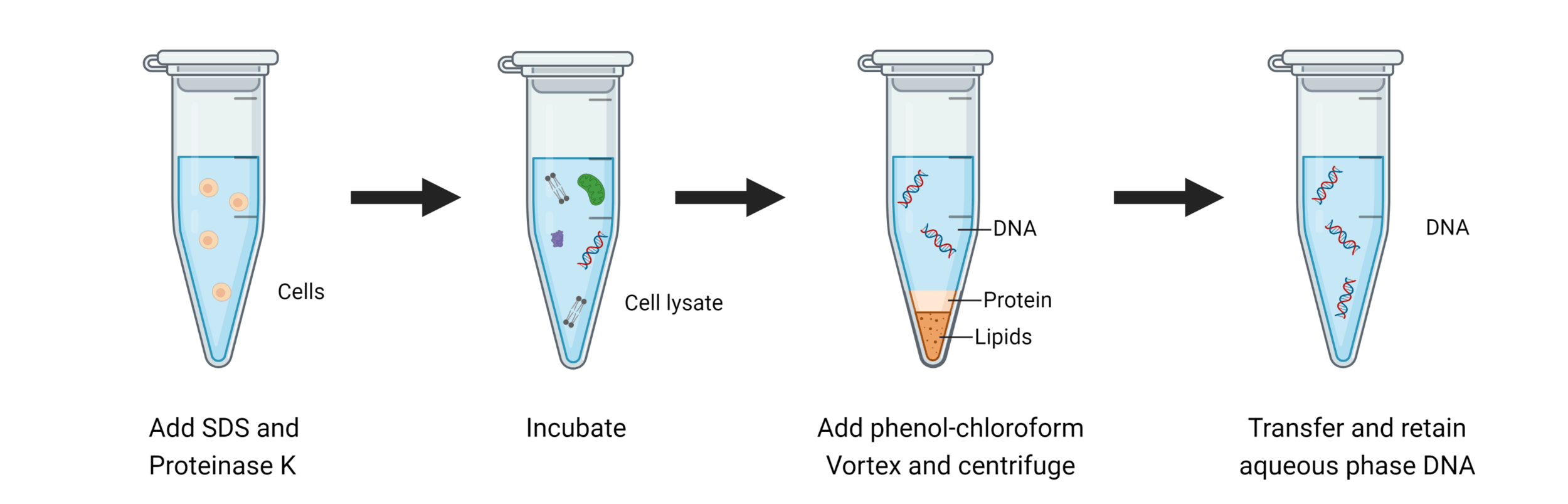
DNA extraction
Computationalists need to understand the underlying source of our data for quality control
Fun fact: Pitt has a beer brewing class (ENGR 1933)
We let our bacterial culture produce our products of interest
Biotechnology frequently uses massive E. coli cultures to produce bioproducts
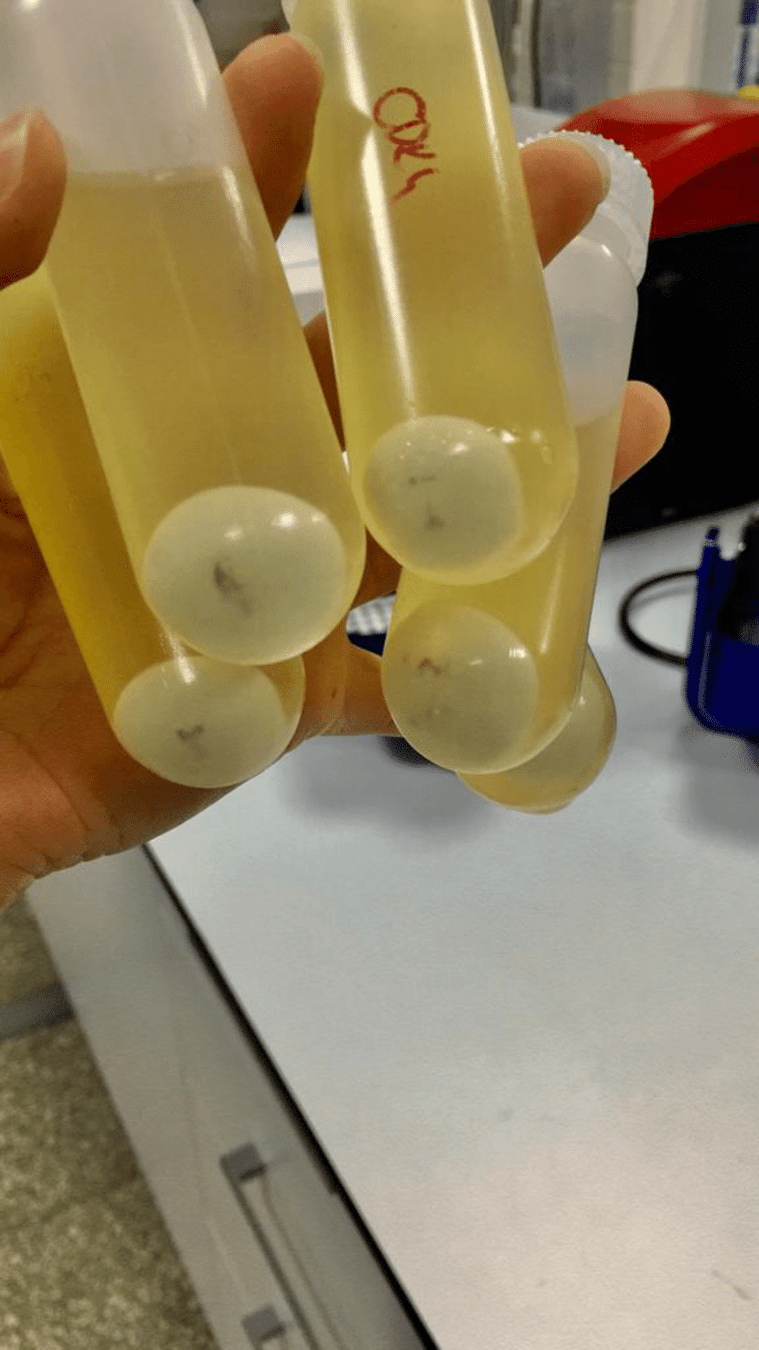
Great! We have our cells, but how can we get DNA out of our cells?
The first step is always to centrifuge and separate our cells and media
Keep the part that has our component of interest (DNA)
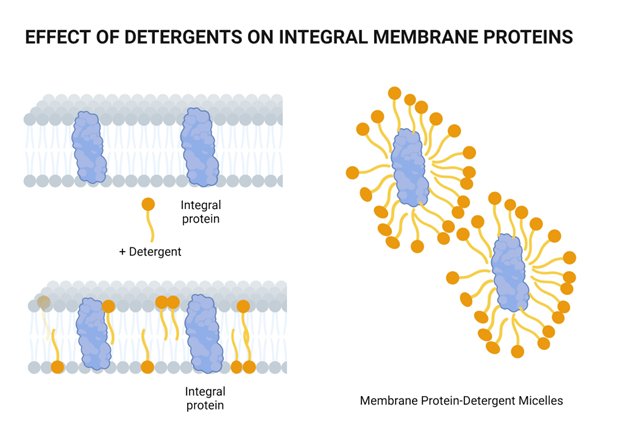
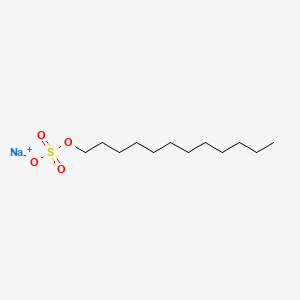
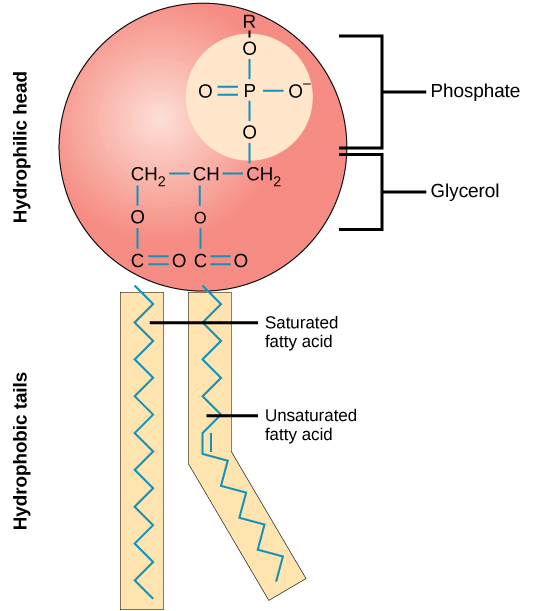
Chemical lysis destabilizes the lipid bilayer and denatures proteins
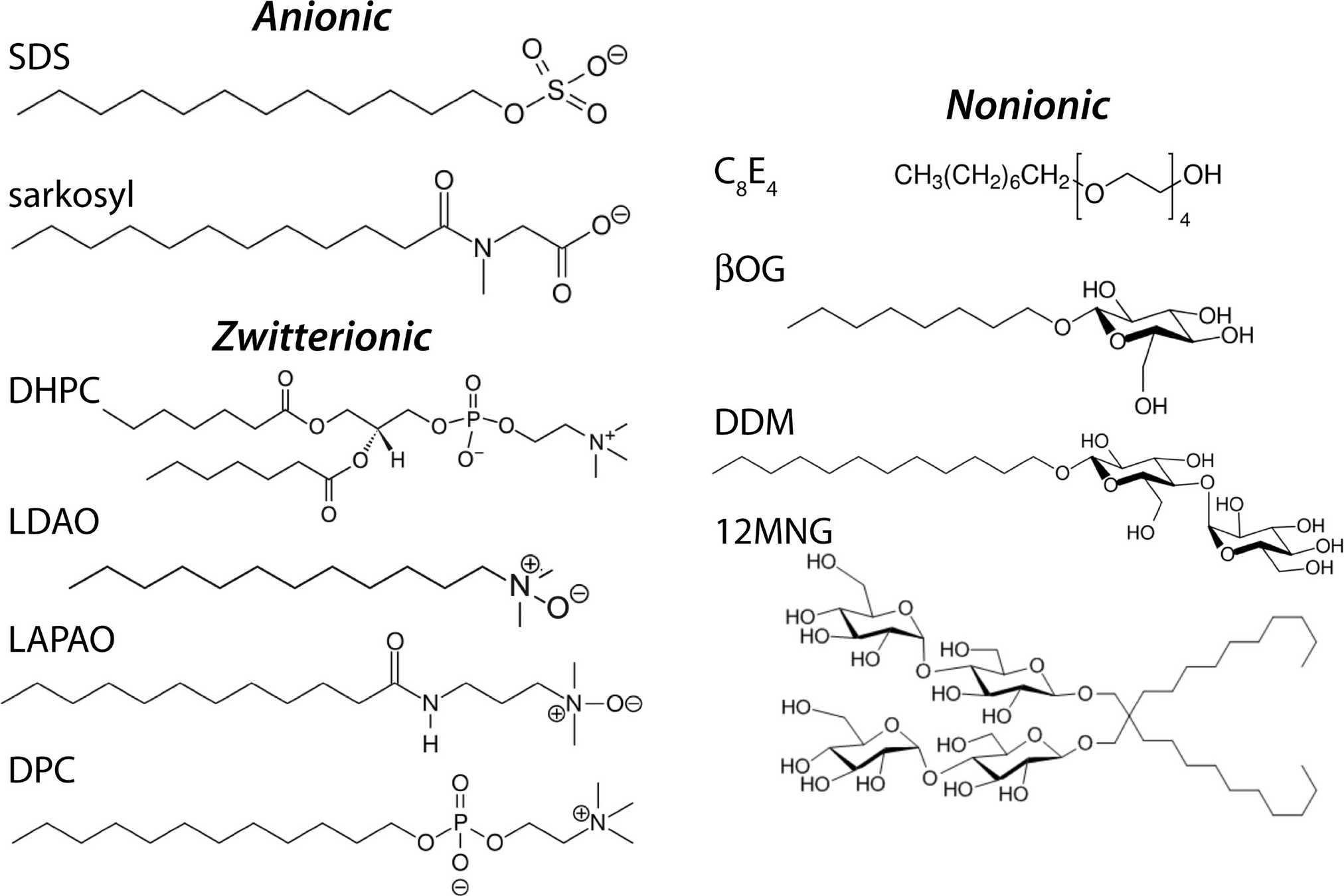
Surfactants have a hydrophilic head and hydrophobic tail
What's the primary difference, and how does this change its behavior?
Surfactants possess a single hydrophobic tail. Why does the incorporation of these surfactants destabilize the phospholipid membrane?
Please note: TopHat questions are ungraded. Engaging honestly with the question will benefit you far more than any shortcuts.
DNA purification
We need to exploit physicochemical property differences (such as solubility, charge, and hydrophobicity) to separate DNA from other biomolecules
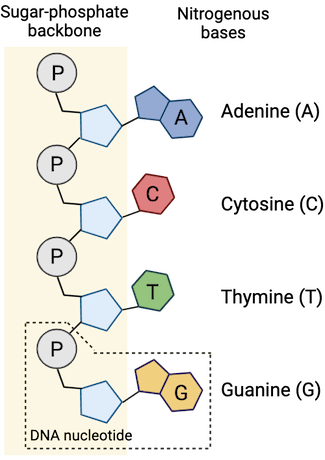
Phosphate backbone
(negative charged)
Denatures and aggregates at interface
Phenol
Chloroform
Water
Nonpolar
DNA
RNA
Protein
Lipids
Collecting our aqueous phase selects only DNA and RNA
Under high-salt conditions, negatively charged DNA binds to the positively charged silica membrane via electrostatic interactions
Contaminants like proteins and salts do not bind or are washed away
DNA is then eluted with a low-salt buffer or water
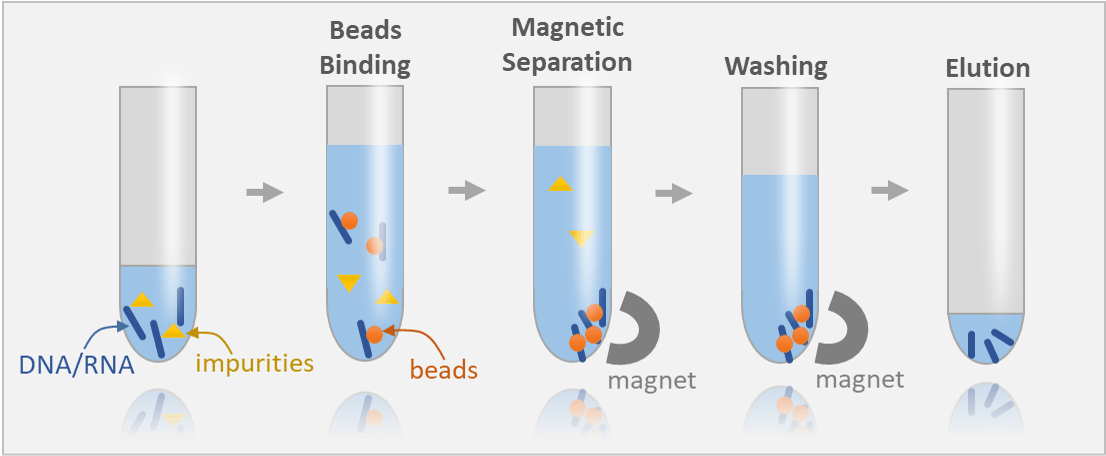
Magnetic beads coated with DNA-binding agents (e.g., silica or polymer) selectively adsorb DNA in the presence of binding buffers
Magnetic fields are used to separate beads with bound DNA from the solution, allowing for washing away impurities like proteins, RNA, and salts
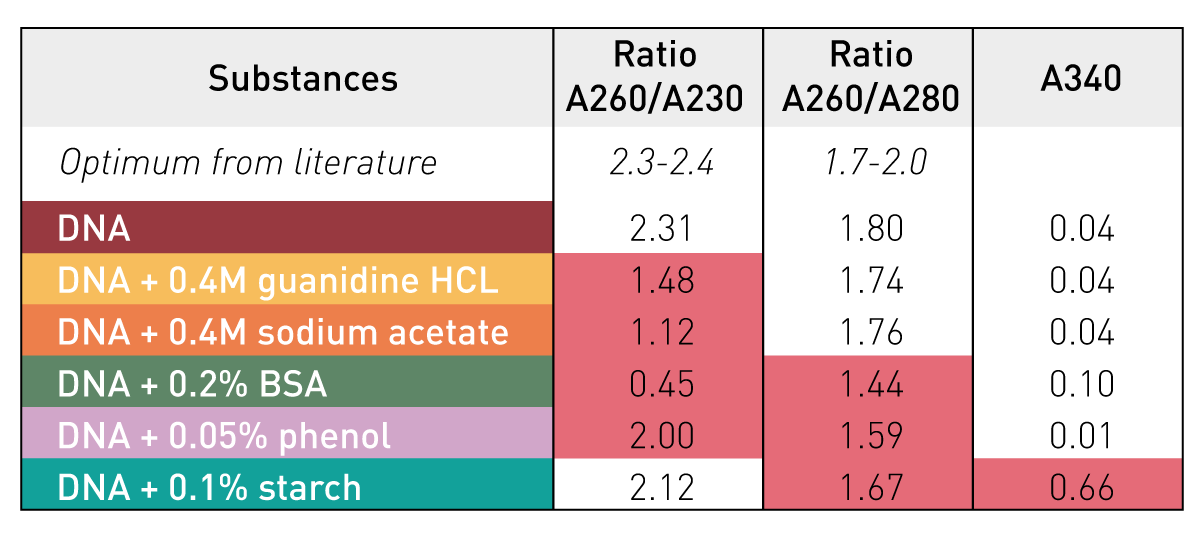
DNA quality quantification
DNA
Likely contaminants
RNA contamination can inflate DNA quantification readings due to similar properties
RNA
Protein
Why it's a problem
Proteins can inhibit enzymatic reactions in library preparation and distort DNA quantification
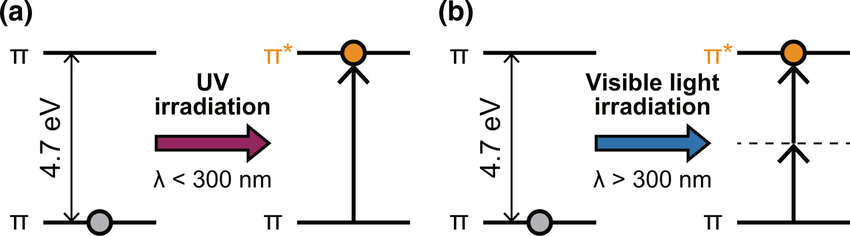
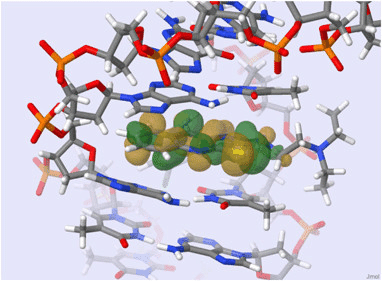
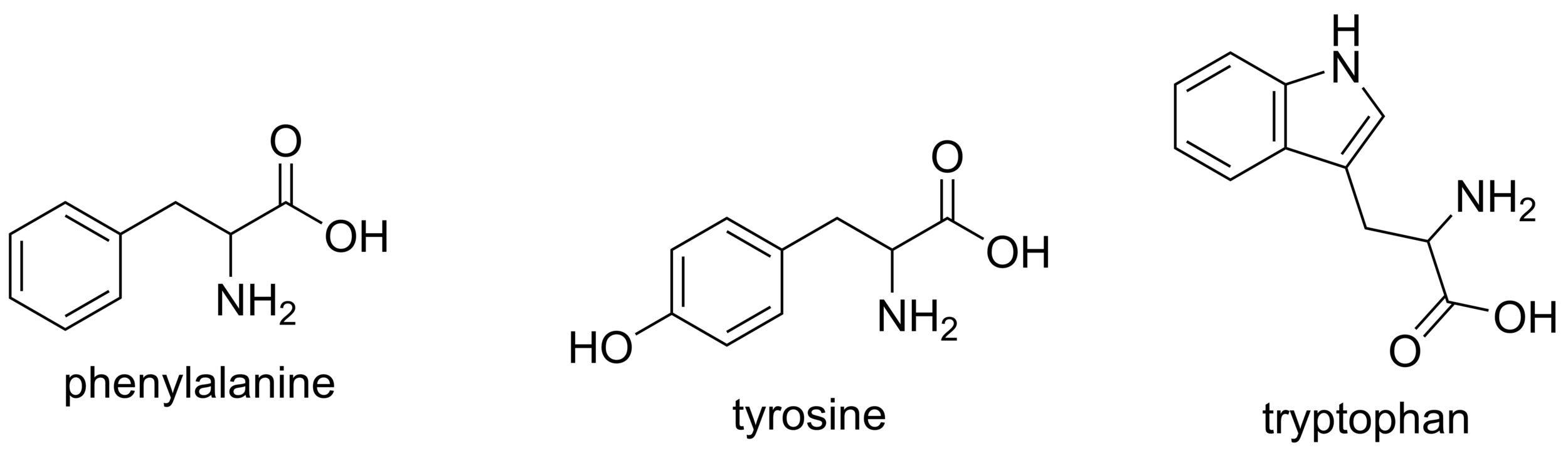
Molecules with aromatic rings absorb UV light strongly due to their conjugated π-electron systems
UV light excites electrons in the π-bonds of aromatic systems to higher energy states
Proteins absorb UV light primarily at 280 nm, mainly due to aromatic amino acids
DNA and RNA absorb UV light at 260 nm because their bases contain highly conjugated double bonds
A260/A280 ratio relates to sample purity
Methods include
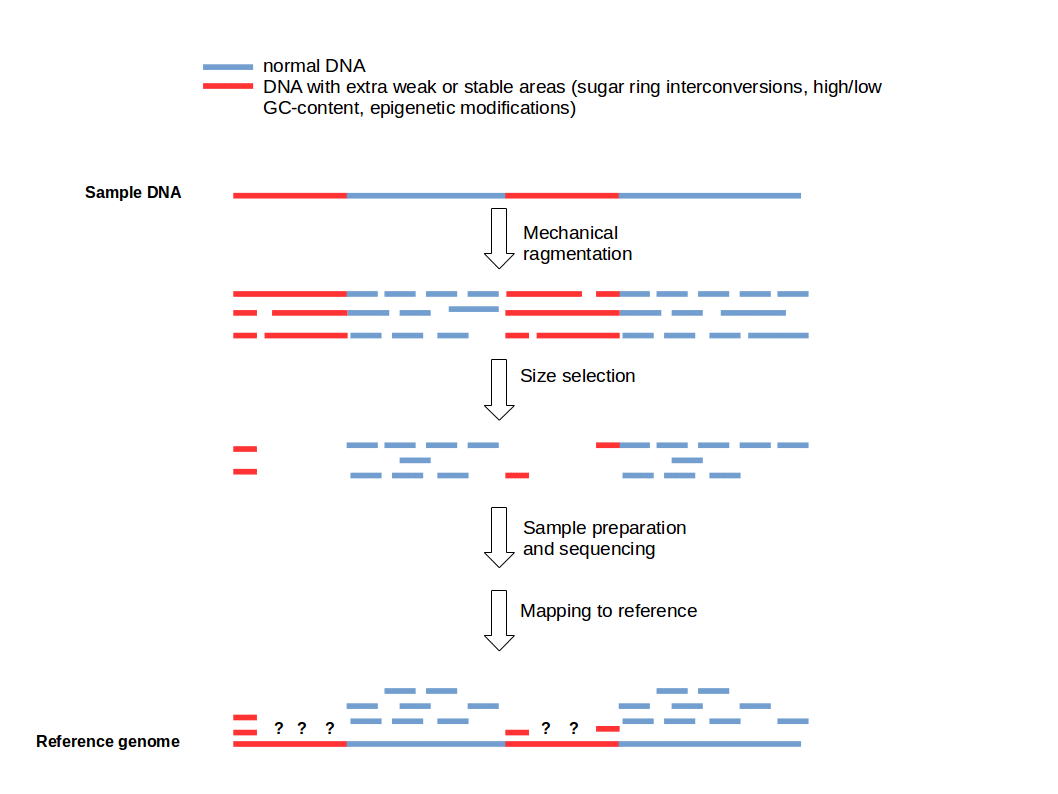
Long DNA molecules cannot be sequenced by most platforms due to size constraints
DNA is fragmented to an optimal size range (e.g., 200–500 bp) for efficient sequencing and alignment
Adapters are short, synthetic DNA sequences that are ligated to the ends of DNA fragments during library preparation
During next-generation sequencing library preparation, short “adapter” sequences are added to the ends of DNA fragments. Which of the following best describes the primary reason for adding these adapters?
A. To link multiple fragments into a single chain for more efficient sequencing.
B. To selectively remove unwanted DNA fragments before sequencing for a better distribution.
C. To incorporate chemical modifications that prevent secondary structure formation.
D. To provide binding sites for PCR and enable recognition by the sequencing instrument.
Please note: TopHat questions are ungraded. Engaging honestly with the question will benefit you far more than any shortcuts.
All DNA sequencing technologies are designed to produce a distinct signal corresponding to nucleotides in a specific sequence
Common signals
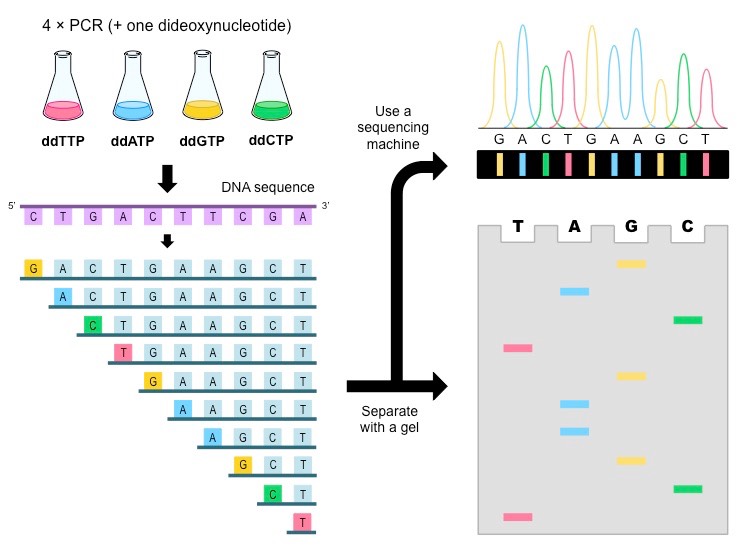
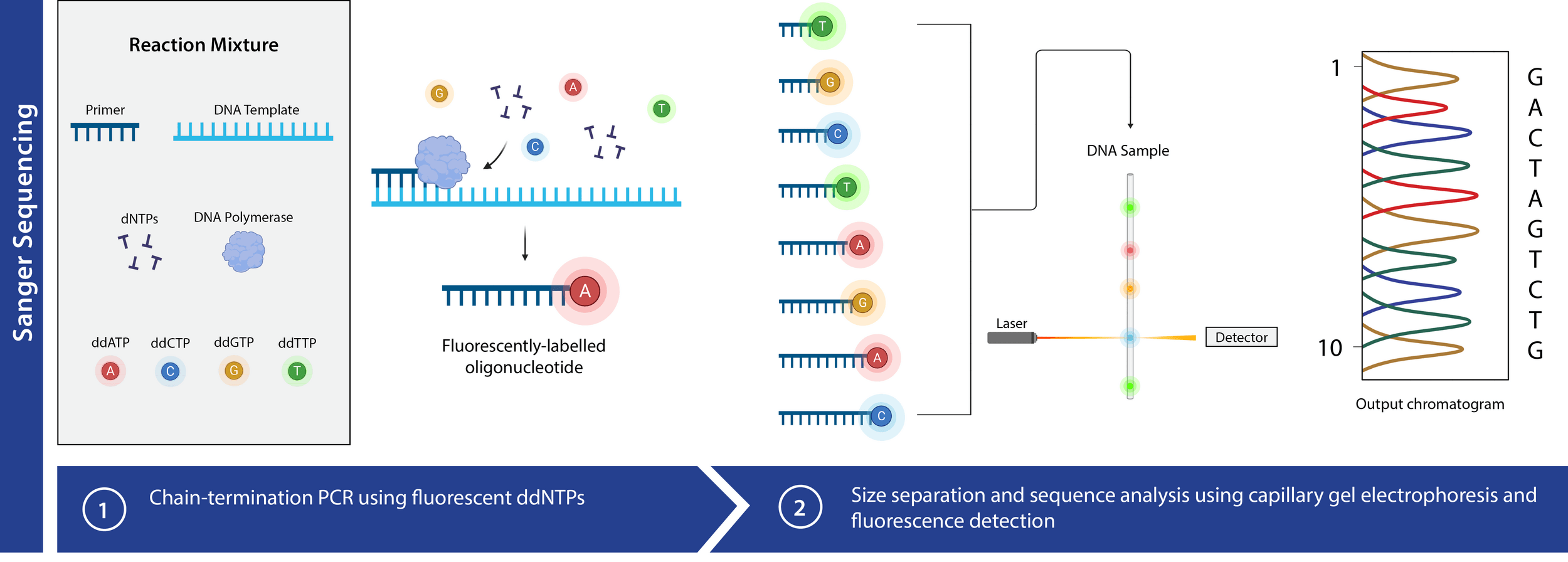
Chain termination (Sanger)
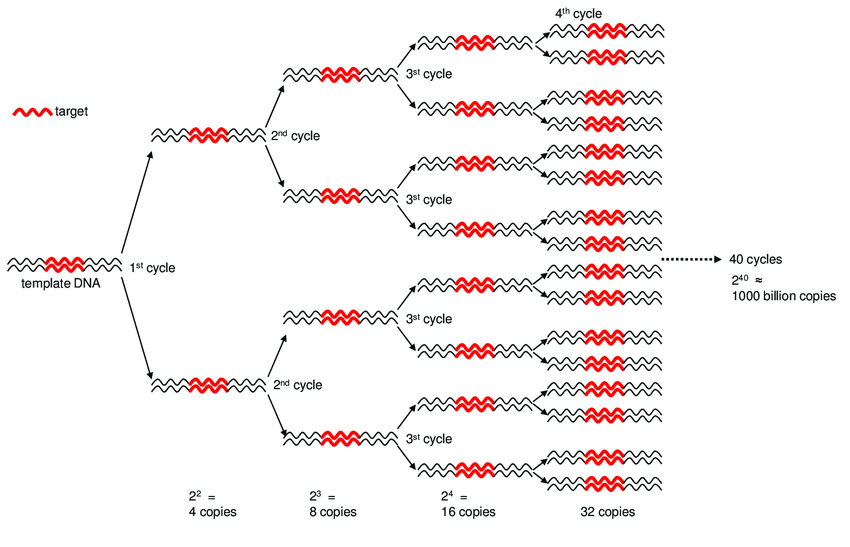
We use DNA polymerase + excess nucleotides to make copies of DNA
When excited by light, fluorescent tags emit distinct signals, providing a mechanism to detect nucleotide identity
Issue: How can we determine where the signal is coming from in the sequence?
The length of a DNA fragment can be used to specify a nucleotide location (i.e., the last nucleotide)
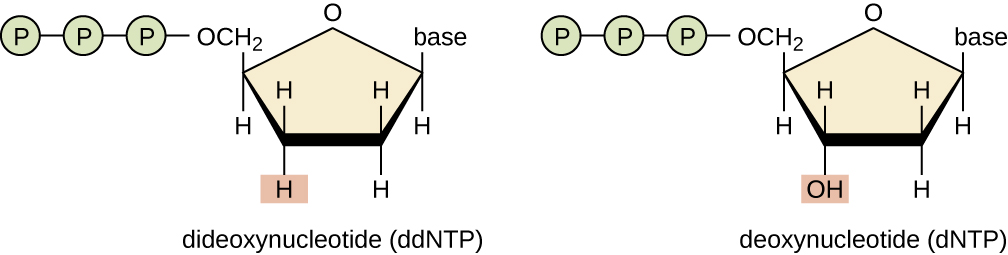
What happens if we don't have the 3' OH?
We cannot add another nucleotide
We will be left with DNA strands of variable length with an optical-based signal at the end
When DNA polymerase adds a
ddNTP
, it cannot add any other
nucleotide
Ratio is usually
1
:
100
Variable-length fragments
Fragments sorted by length
Last nucleotide order
Why would we need separate beakers?
Once we have fragments, how can we separate them by length?
Gel electrophoresis!
Cannot differentiate between radioactive nucleotides
Only need one PCR!
Capillary gel electrophoresis can accelerate fragment length sorting and detection
Unique fluorescence signal per ddNTP produces a chromatogram
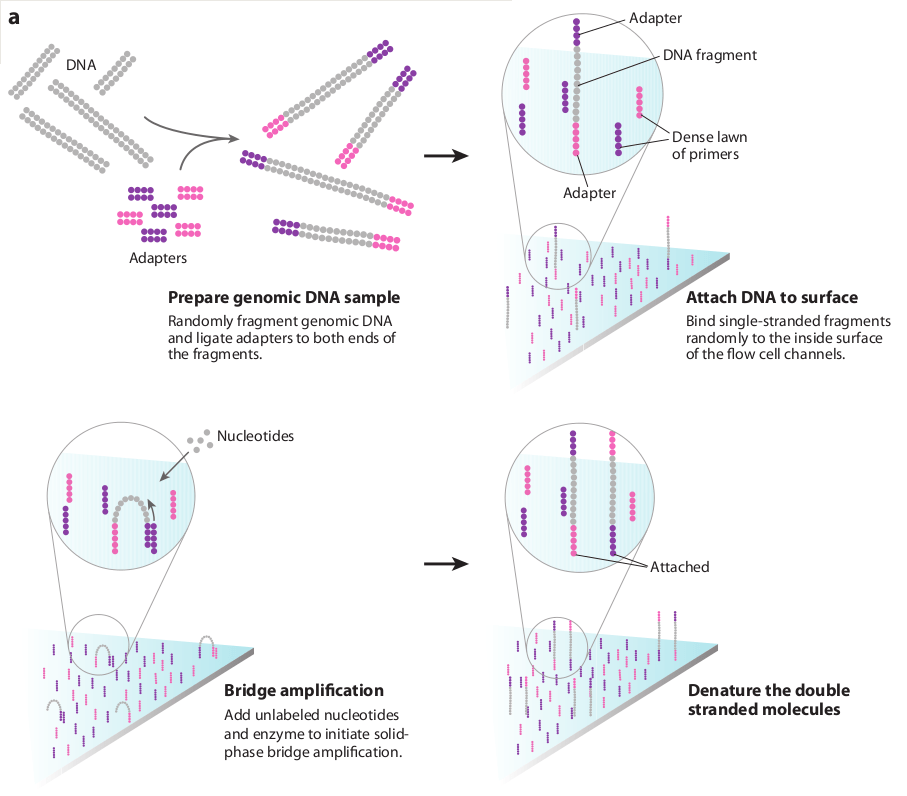
Sequencing by synthesis (Illumina)
What if we could identify nucleotides as they are being added, allowing us to sequence faster and at a larger scale?
Bridge amplification creates double-stranded bridges
Clusters will give off a stronger signal compared to a single fragment
Double-stranded clonal bridges are denatured with cleaved reverse strands
Even with immobilization, the signal from a single fragment is often too weak to detect
Forward
Reverse
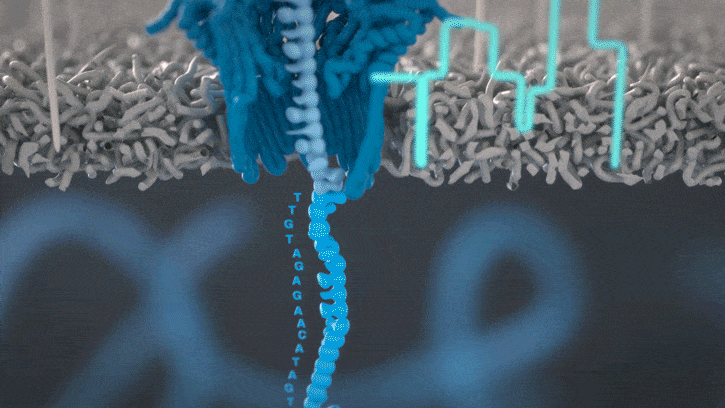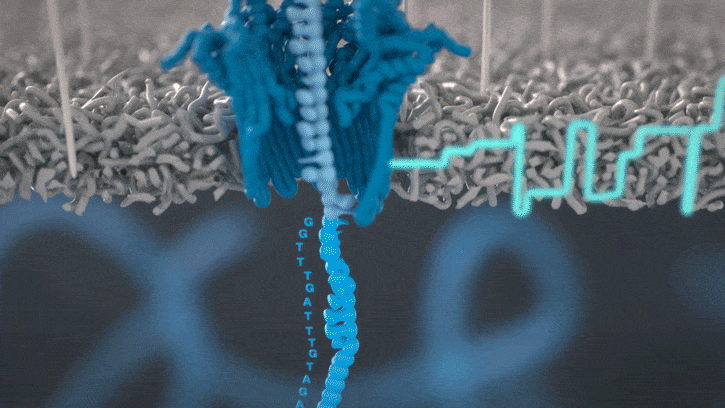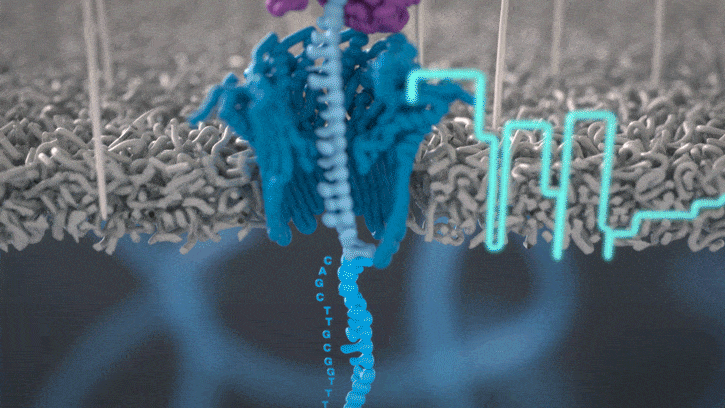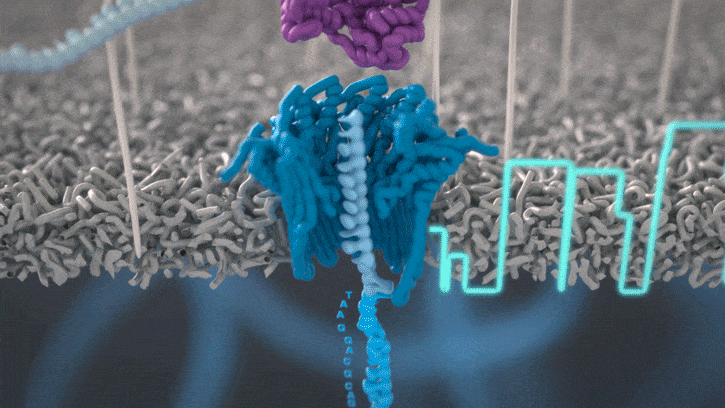
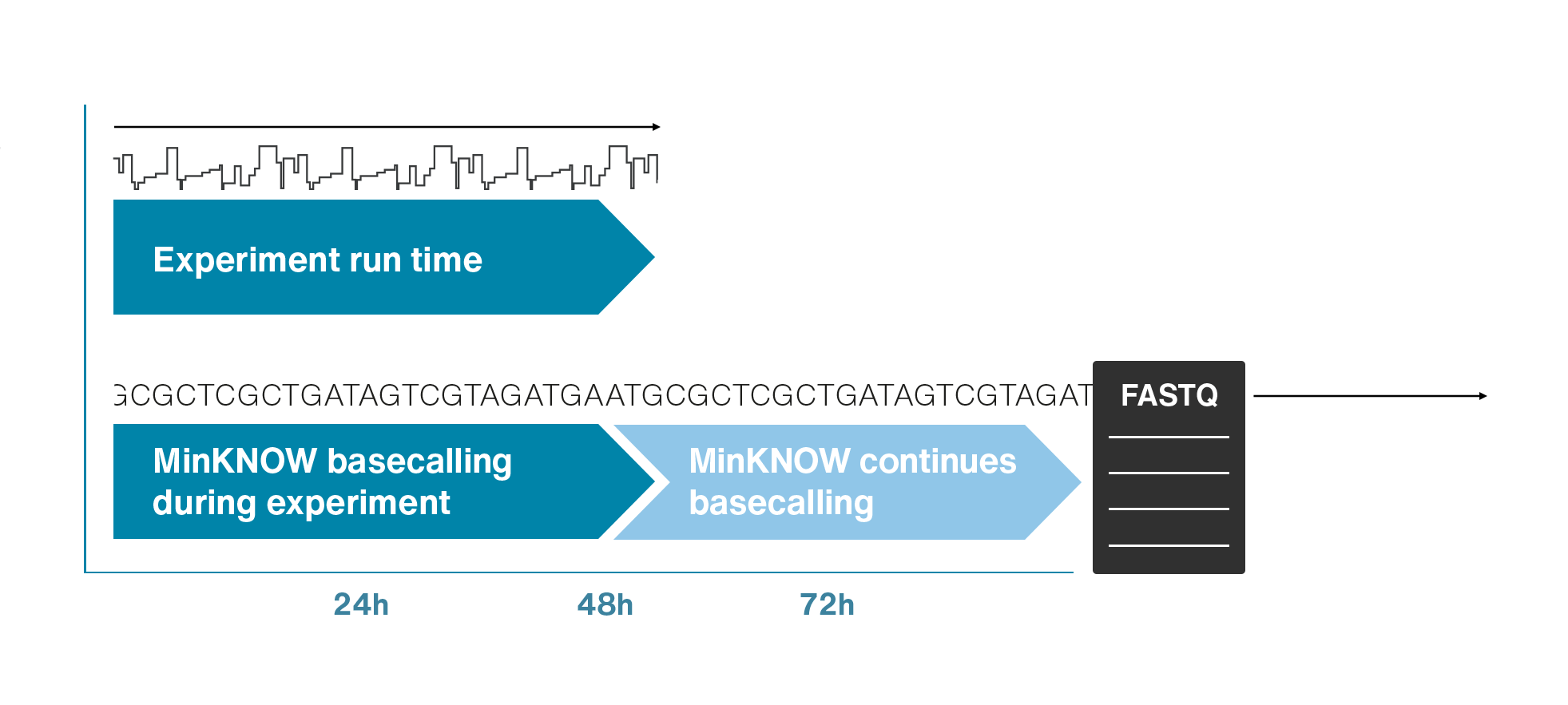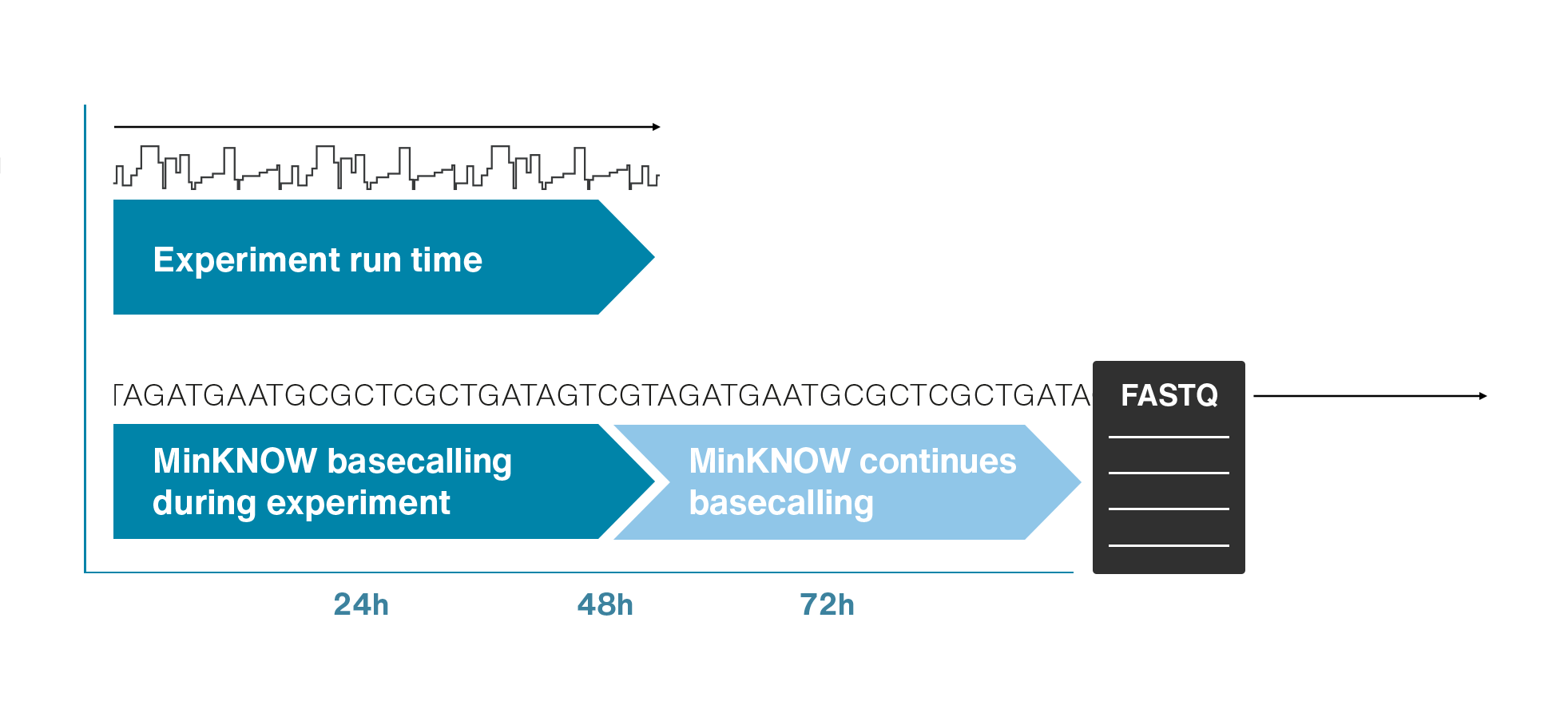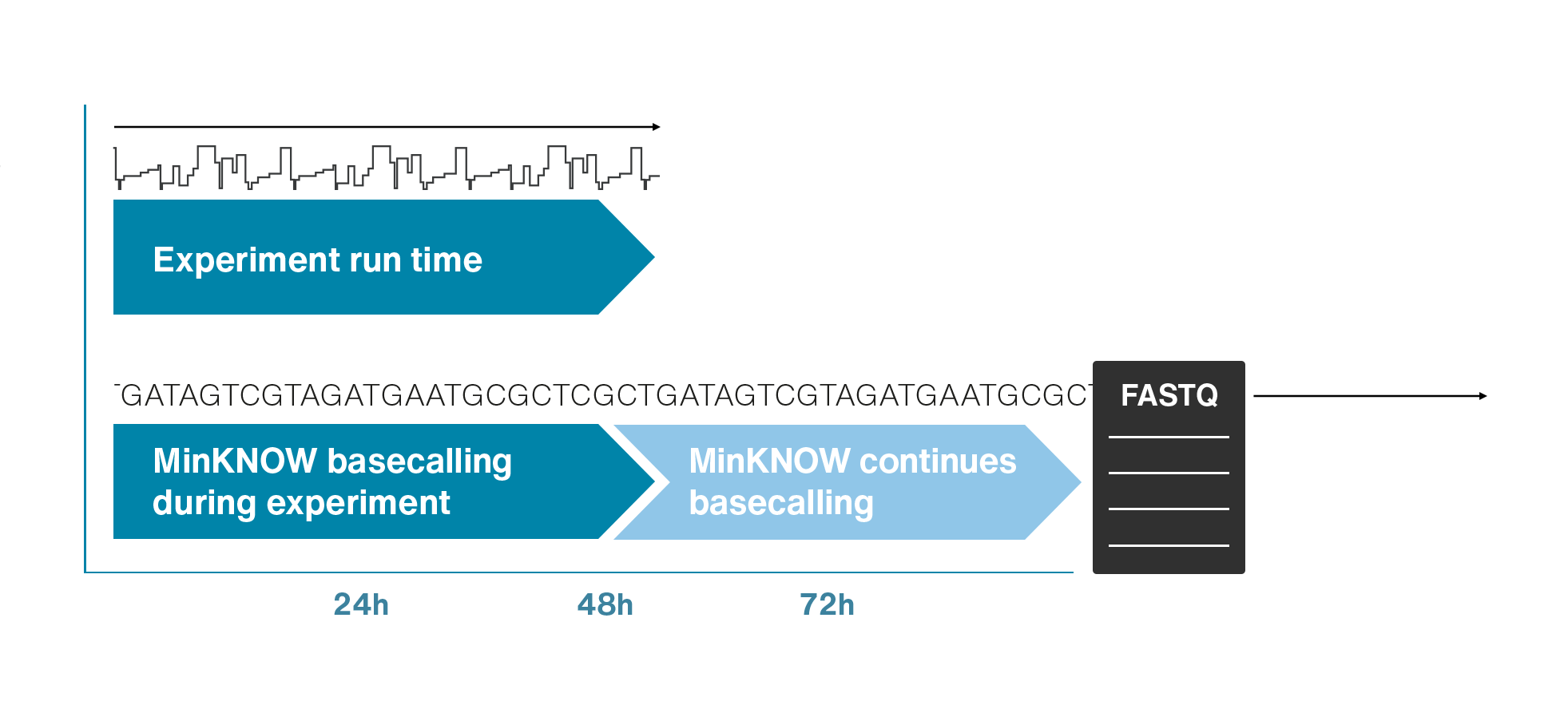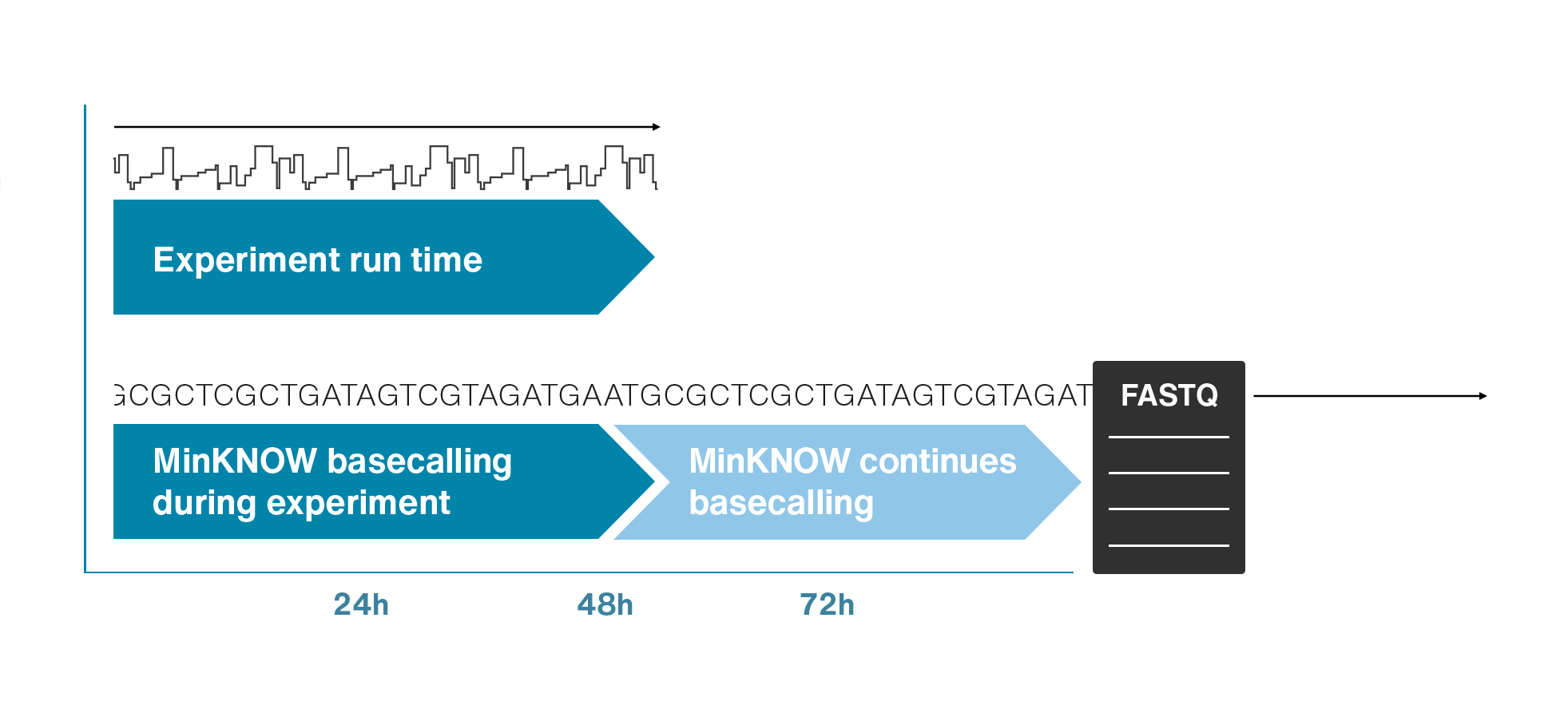
Single molecule sequencing (Nanopore)
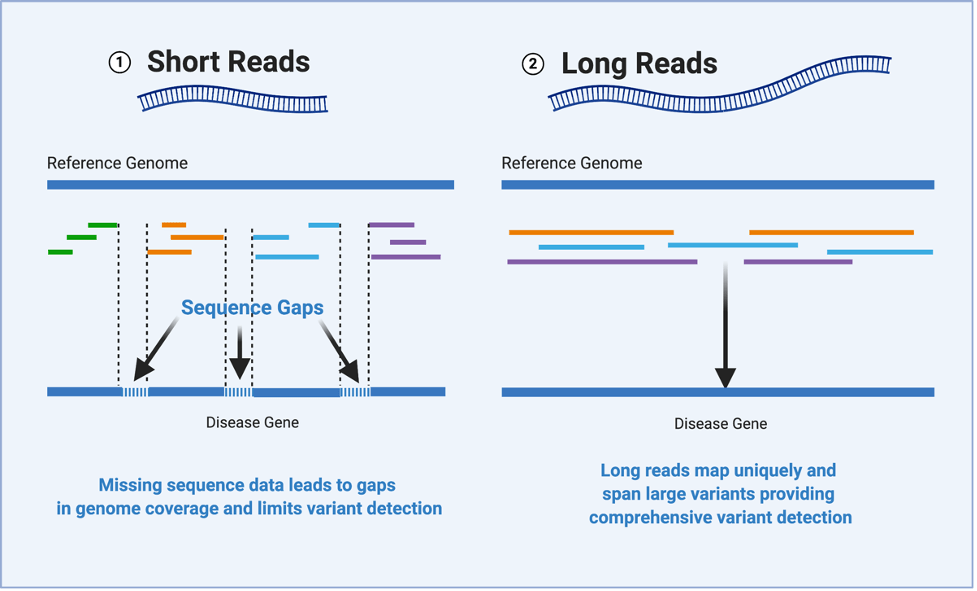
Short DNA reads make genome assembly difficult, especially in repetitive regions
Nanopore sequencing detects nucleotide sequences by measuring changes in ionic current as DNA passes through a pore
Match each modern sequencing technology with the correct combination of features or characteristics.
Please note: TopHat questions are ungraded. Engaging honestly with the question will benefit you far more than any shortcuts.
Lecture 02B:
DNA sequencing -
Methodology
Lecture 02A:
DNA sequencing -
Foundations
Today
Thursday