Estimating effects of mutations to SARS-CoV-2 proteins from natural sequences
Jesse Bloom & Richard Neher
Fred Hutch Cancer Center / HHMI

Determining effects of viral mutations is important
- Interpret consequences of mutations seen during viral surveillance.
- Inform design of drugs to target constrained regions.
- Understand function and mechanisms of viral proteins.
Traditional way to determine effect of mutations is experiments
Traditional way to determine effect of mutations is experiments
Traditional way to determine effect of mutations is experiments
Traditional way to determine effect of mutations is experiments
Traditional way to determine effect of mutations is experiments
My group tries to do such experiments at large scale via deep mutational scanning

Yeast display or lentiviral pseudotype libraries allow us to measure many mutants at once by pooling them all together and reading out effects of mutations by deep sequencing (Starr et al, 2020; Dadonaite et al, 2022)
Limitations of using experiments to understand mutation effects
- Laborious in three years, entire field has only made large-scale measurements for two proteins:
- spike and its RBD (Starr et al, 2022; Dadonaite et al, 2022)
- Mpro (Flynn et al, 2022; Iketani et al, 2022)
- Lab assays measure effects of mutations in cells or mice, not humans.
- Some viral proteins have poorly understood functions that lack good lab assays
Nature is "testing" effects of viral mutations in humans all the time
Average neutral single-nucleotide mutation has occurred ~15,000 independent times in human transmitted SARS-CoV-2
- Viral substitution rate at synonymous sites: ~7.5e-4 substitutions/year (Neher, 2022)
- Typical infection duration: ~5 days = 0.01 years/infection
- Total human infections with SARS-CoV-2: ~6e9 infections (as of early 2023)
- So total synonymous substitutions per site: 7.5e-4 x 0.01 x 6e9 = 45,000
- There are three possible mutations per site: 45,000 / 3 = 15,000
- Mutation spectrum uneven, so some mutations have occurred more than others:
- C->T mutations have occurred ~50,000 times
- A->C mutations have occurred ~1,000 times
We can use publicly available human SARS-CoV-2 sequences to "read out" effects of viral mutations on human transmission
- We use the ~6.5 million public sequences in the UShER mutation-annotated tree
- These sequences represent ~0.1% of all human SARS-CoV-2 infections as of early 2023
First calculate how often each mutation expected to be observed without selection by analyzing 4-fold degenerate sites
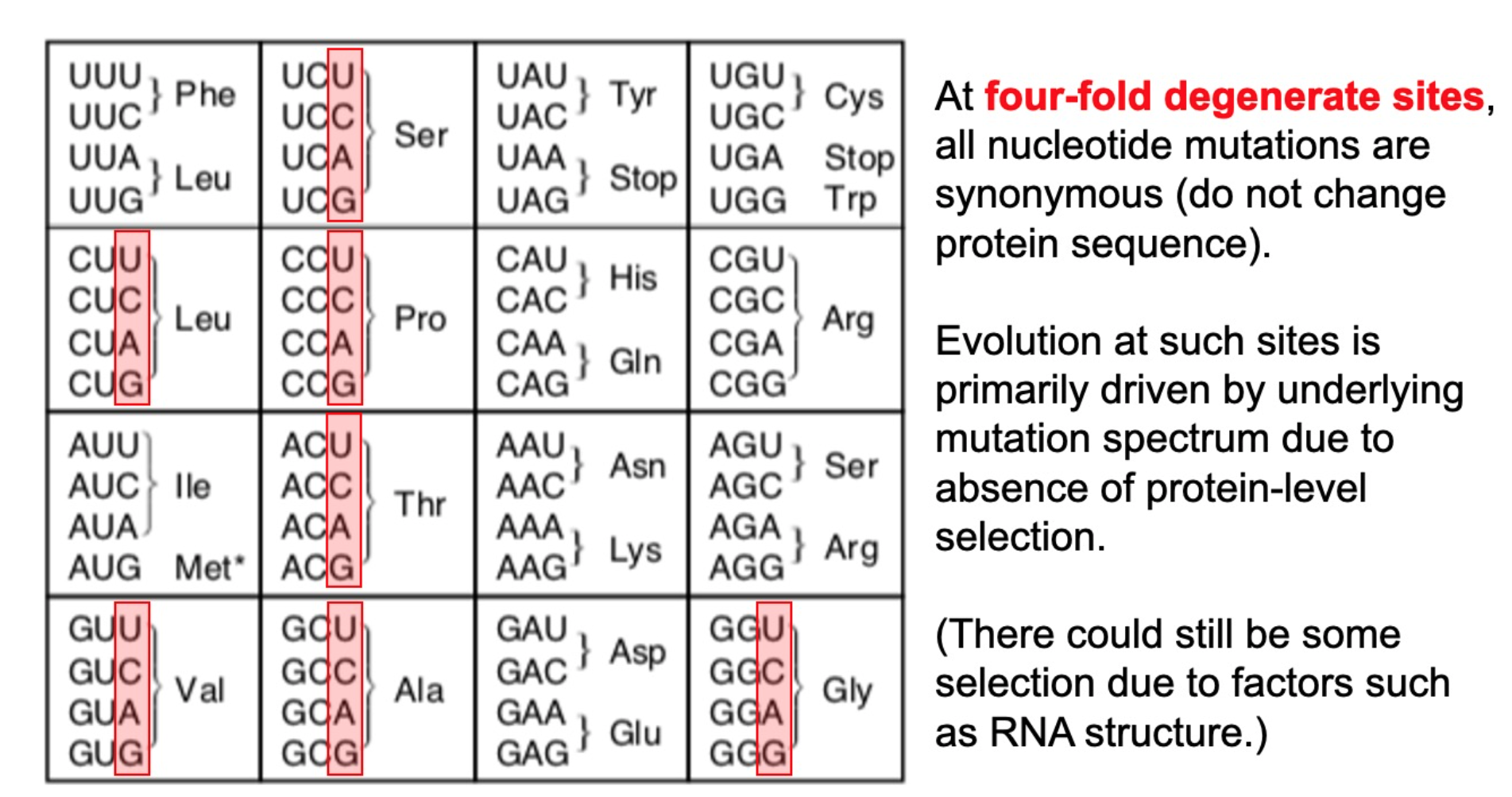
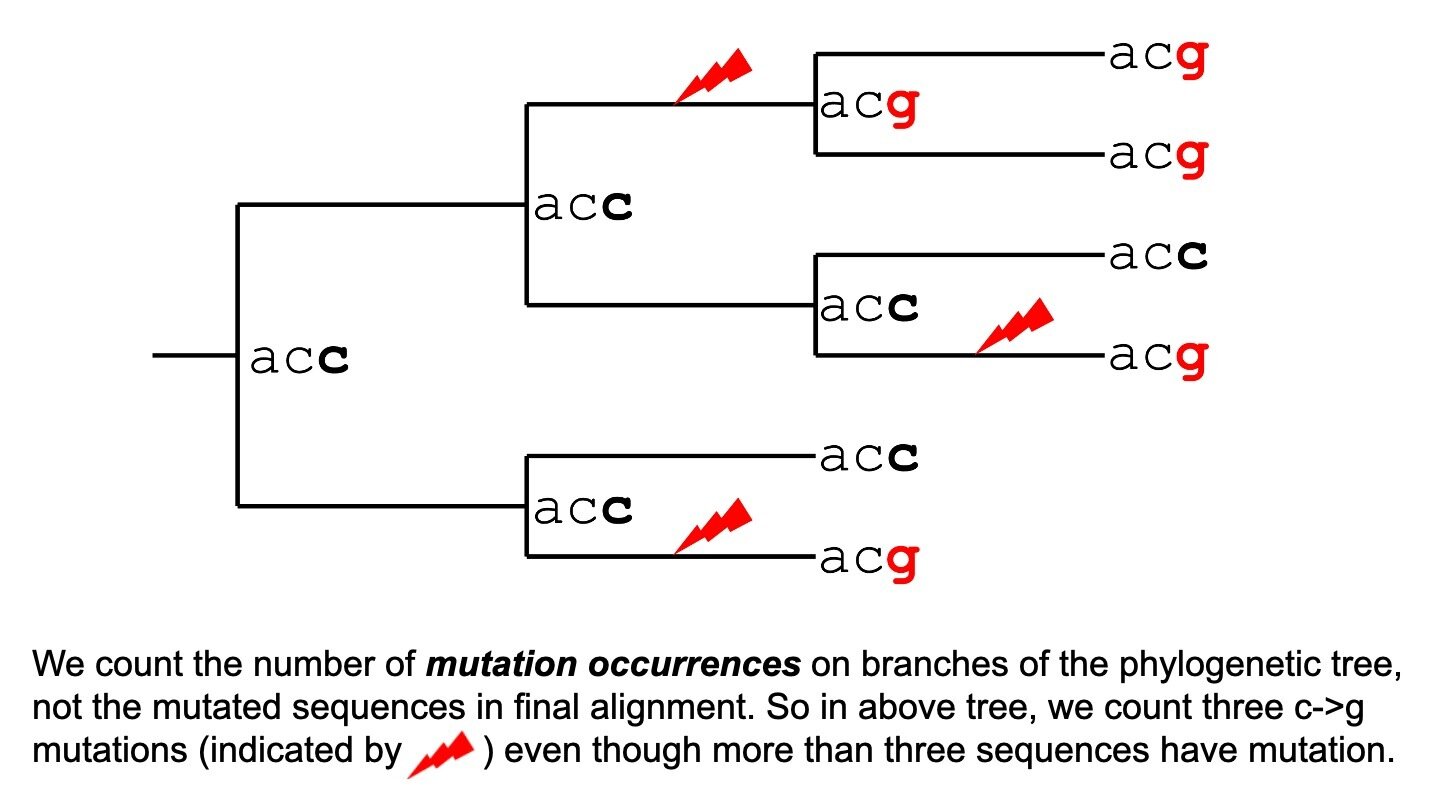
We count unique occurrences of mutation, not number of sequences with mutation
Mutations expected to be observed ~8 to ~500 times in absence of selection
There are enough sequences to calculate effects on a per-mutation basis
- We calculate effect as log of actual versus expected mutation counts
- Effects of zero indicate neutral mutation, negative indicates deleterious mutation
- Estimates are more accurate (less noise) for mutations with larger expected counts
Distribution of effects of all mutations
We can see which genes are under strong purifying selection
Among accessory genes, ORF3a is under strongest selection against stop codons
Experiments show that only accessory gene deletion that strongly attenuates virus in animal models is ORF3 (McGrath et al, 2022)
We can also look in detail at mutation level
These maps can identify constrained sites
Estimated mutation effects are robust to sequence sampling location
Estimated mutation effects are robust to viral clade identity
Estimated mutation effects correlate well with deep mutational scanning
Two spike deep mutational scans using different underlying methodologies: lentiviral pseudotyping of spike or yeast display of RBD