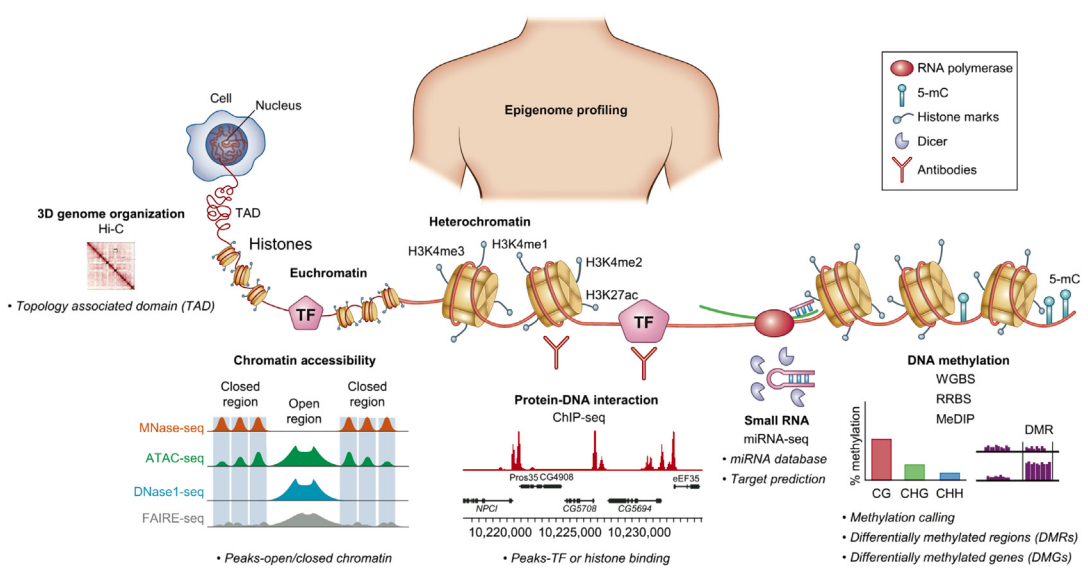
Introduction to epigenetics and ATAC-Seq
Aleksandra Galitsyna
“Analysis of omics data” course
Skoltech
28 February 2022

Sequencing for epigenetics
NGS techniques diversity

Types of binding events in the cell
Proteins and nucleic acids are the most important components of the cell. Their interactions are necessary for functioning and regulation. Types of interactions:
-
DNA-protein interactions, examples:
- replication/reparation/recombination
- transcription
- chromatin modification and folding
-
RNA-protein:
- RNA metabolism regulation
- processing
- translation
- degradation: microRNA, NMD
-
RNA-DNA interactions:
- expression regulation
- chromatin structure maintenance
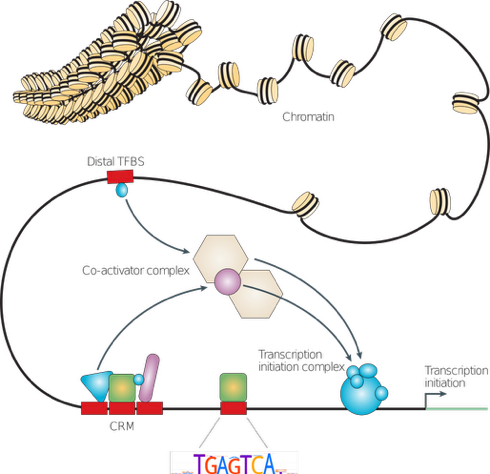
Wasserman & Sandelin, 2004
Types of molecular interactions
-
Direct:
- protein-protein
- RNA-protein
- DNA-protein
- RNA-DNA (R-loops)
-
Indirect:
- Protein-mediated
- RNA-mediated
-
Colocalization:
- Compartments
- Hubs
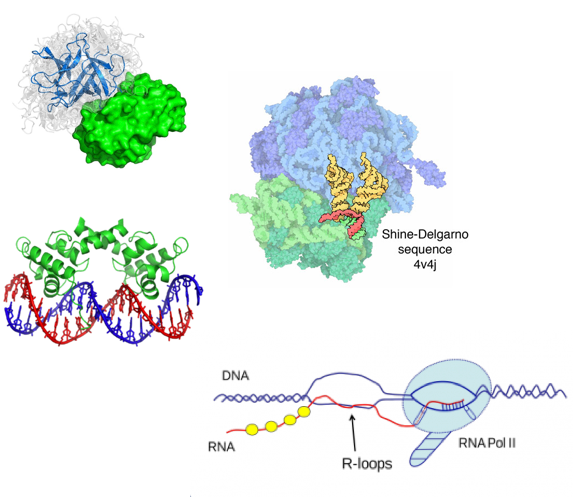
RNA-DNA interactions
Engreitz et al. 2016
ChIP-Seq
Binding of protein is typically specific to DNA sequence:

Binding pattern
DNA-protein binding
List of DNAs
with binding events
courtesy of Ivan Kulakovsky, Pavel Mazin
ChIP-Seq
Chromatin-immunoprecipitation followed by sequencing:
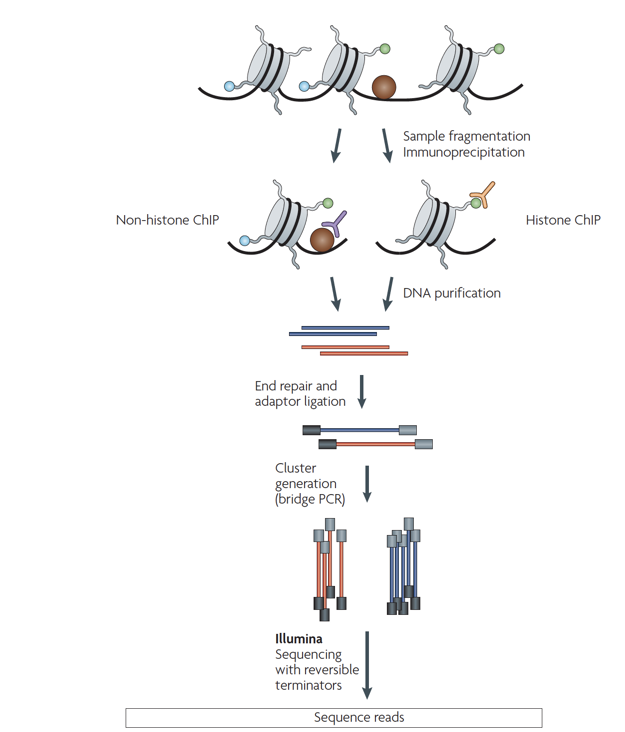
ChIP-Seq: input experiment
Unwanted factors contributing to sequencing & processing output:
- DNA accessibility
- DNA amplification efficiency
- Mappability
"Input" experiment is a ChIP-Seq control without precipitation step. It is used for normalisation of ChIP-Seq.
Park, Nat Reviews Genetics, 2009
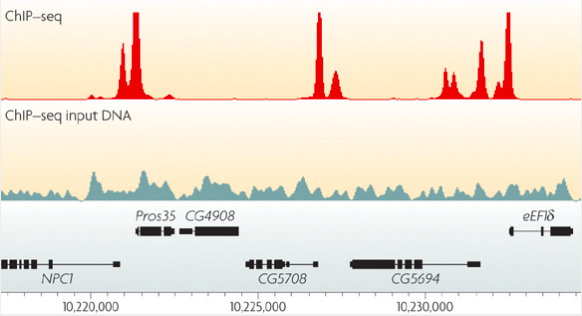
ChIP-Seq: peak calling
In order to predict binging events we need to call peaks in ChIP-Seq data:
Mahony & Pugh 2015
ChIP-Seq: binding pattern

- As an output from ChIP-Seq we have a list of peaks.
- In theory, each peak should have a binding event.
- Imagine you have the peaks with the following sequences:
- What is the binding pattern?
courtesy of Ivan Kulakovsky, Pavel Mazin
ChIP-Seq: binding pattern
- Seems to be easy to find the binding pettern:

Question: Can you propose a computational approach to find it?
courtesy of Ivan Kulakovsky, Pavel Mazin
ChIP-Seq: motifs search
-
Let's introduce some variety, which can arise as:
- Suboptimal binding of the protein
- Genome variability
- Errors during the sequencing

courtesy of Ivan Kulakovsky, Pavel Mazin
ChIP-Seq: motifs search
- With possible substitutions, finding the correct pattern is not easy anymore:

We need some advanced algorithms to find binding patterns.
courtesy of Ivan Kulakovsky, Pavel Mazin
Binding pattern representation
- Binding motif is a DNA sequence pattern that has biological significance (e.g. it is a target for protein binding).
- Consider following pattern of sequences:
TATAAT TAAAAT TAATAT TGTAAT TATACT
Consensus is the sequence of the most frequent letters:
T[AG][AT][AT][AC]T
With the consensus sequence, we lose the information:
- about suboptimal binding,
- of background nucleotides frequencies.
courtesy of Pavel Mazin
Binding motif representation
Other representations:
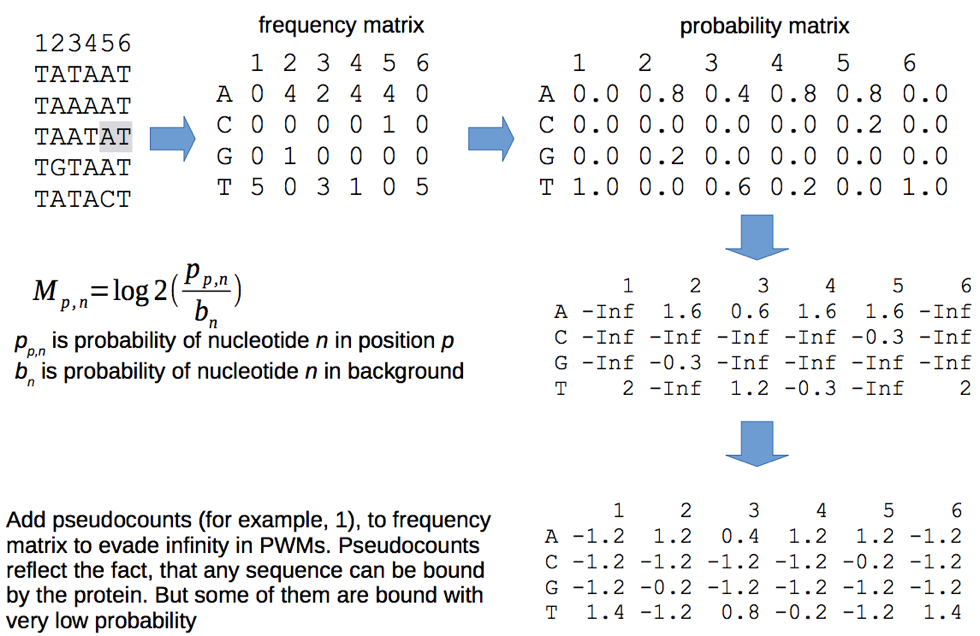
Frequency matrix
Probability matrix
Matrix normalized by background
Position Weight Matrix (PWM)
courtesy of Pavel Mazin
Position Weight Matrix (PWM)

courtesy of Pavel Mazin
Binding motifs search
Approaches:
- Scanning of the motifs from the database
(e.g. JASPAR http://jaspar.genereg.net/ )
Binding motifs search
Approaches:
- Scanning of the motifs from the database
(e.g. JASPAR http://jaspar.genereg.net/ ) - Motifs discovery (de novo search)
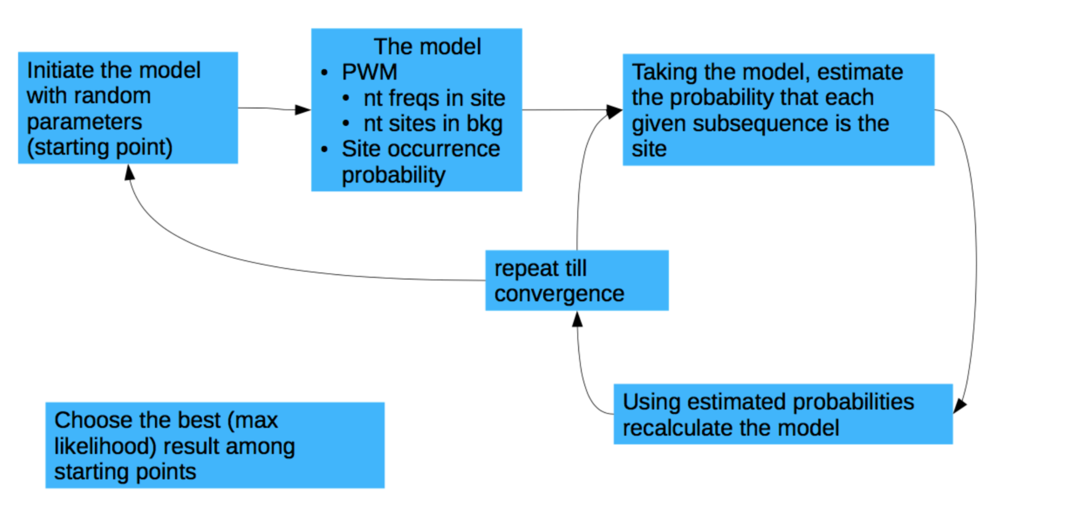
courtesy of Pavel Mazin
ChIP-Seq: motifs search tools
- Web server tools:
-
Command line interface tools (CLI):
- MEME
- HOMER
- Autosome
- ...
-
GimmeMotifs https://gimmemotifs.readthedocs.io/en/master/
- Simple CLI
- Can run multiple other tools
- Databases included
Binding events and DNA motifs
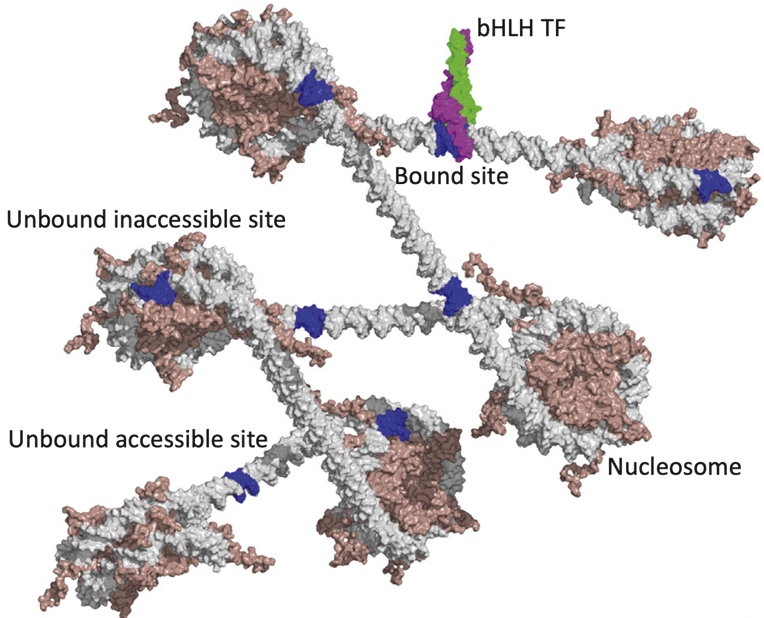
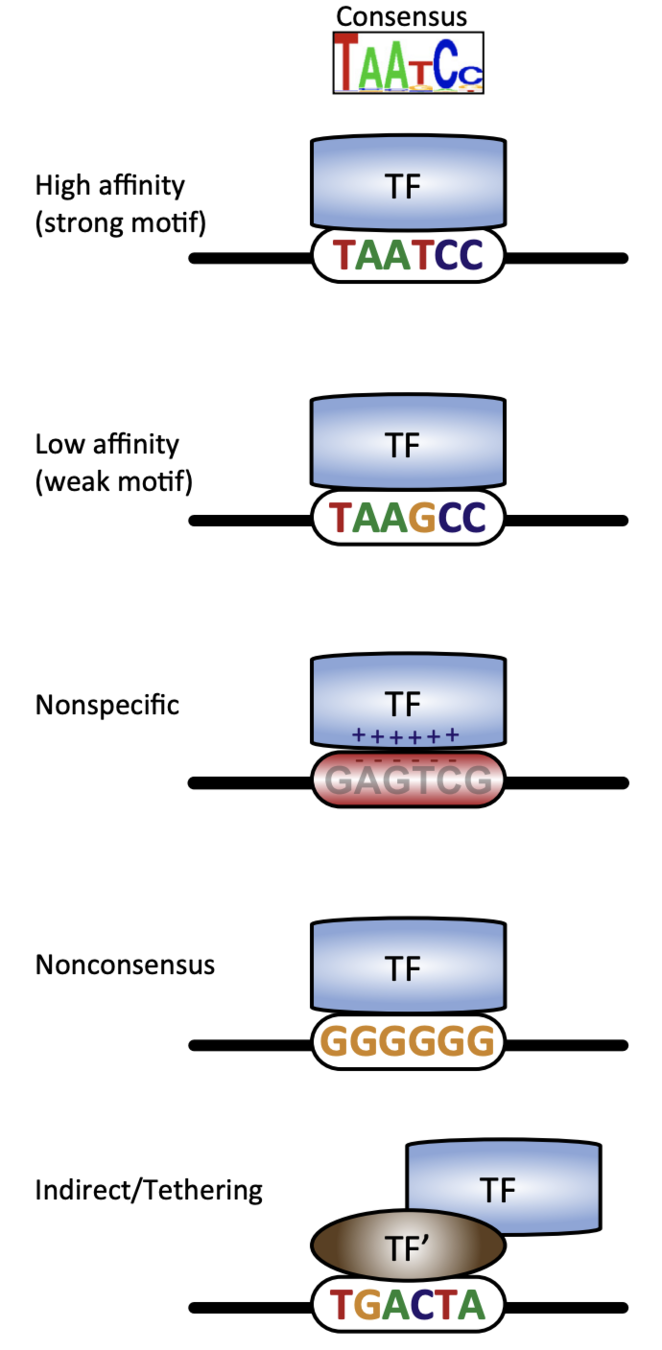
Specific and non-specific binding:
Slattery et al. 2014
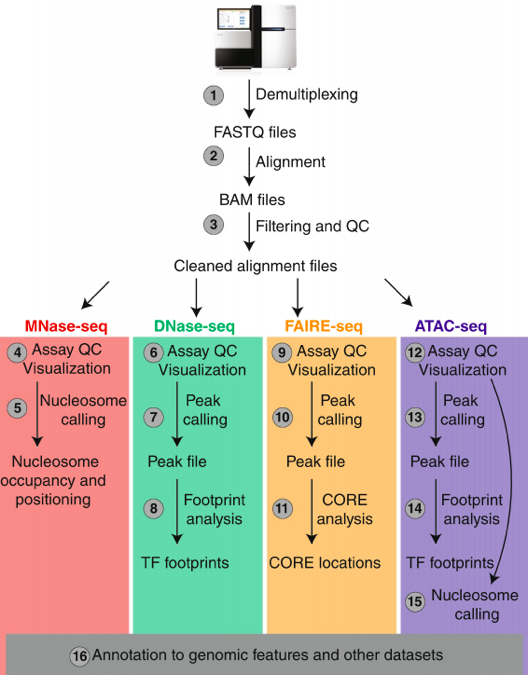
DNase-Seq: experimental protocol
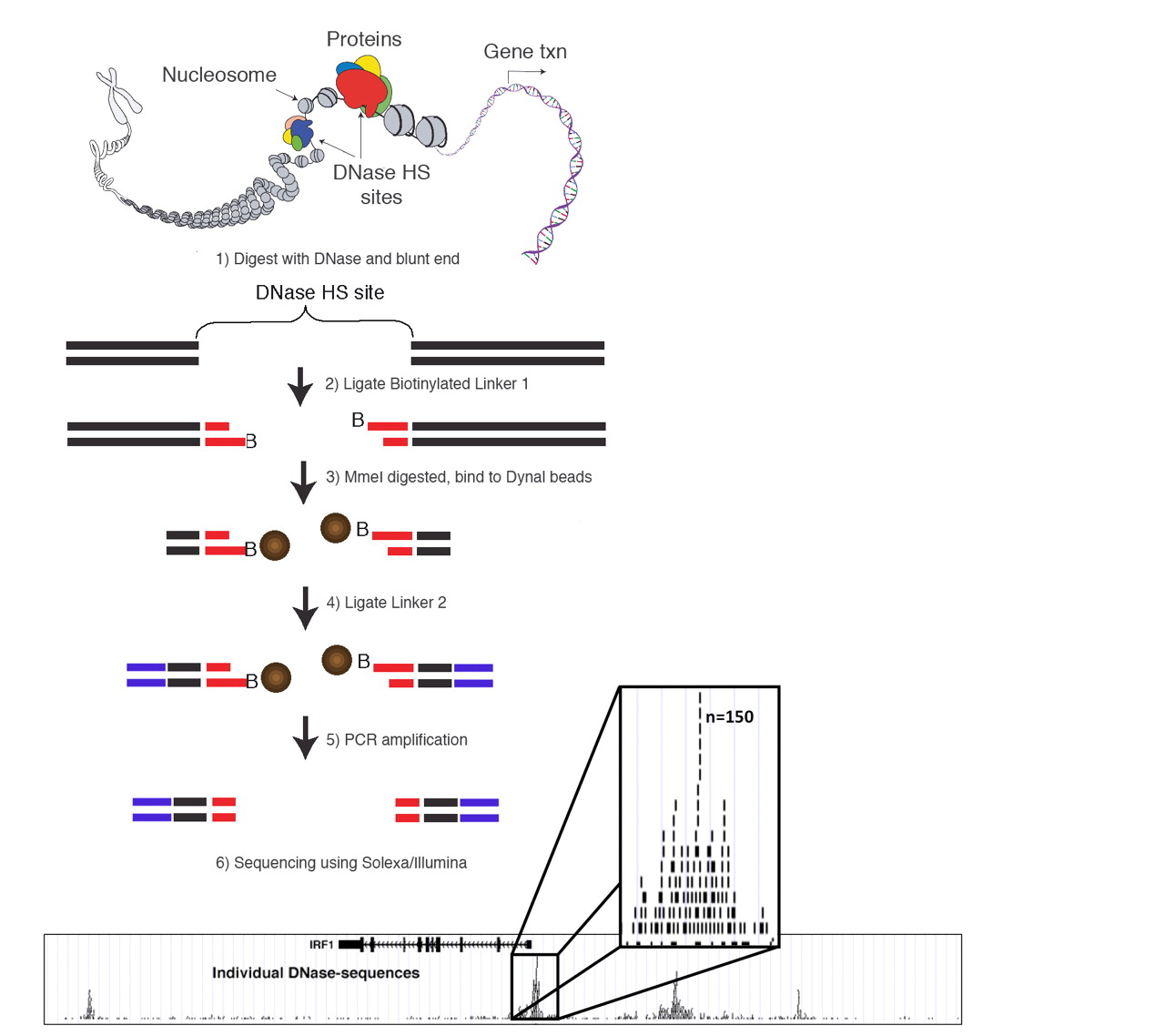
- DNase I hypersensitive sites sequencing
ATAC-Seq: experimental protocol
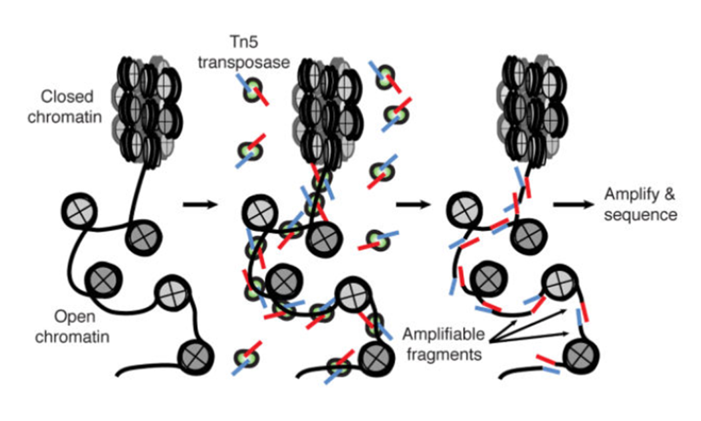
- Assay for Transposase-Accessible Chromatin with high-throughput sequencing
Nucleosome-free regions detection method- Tagmentation: Tn5 transposase cuts DNA and ligates adaptors to open DNA
Buenrostro et al., 2013
"Block-schemes" of Seqs:
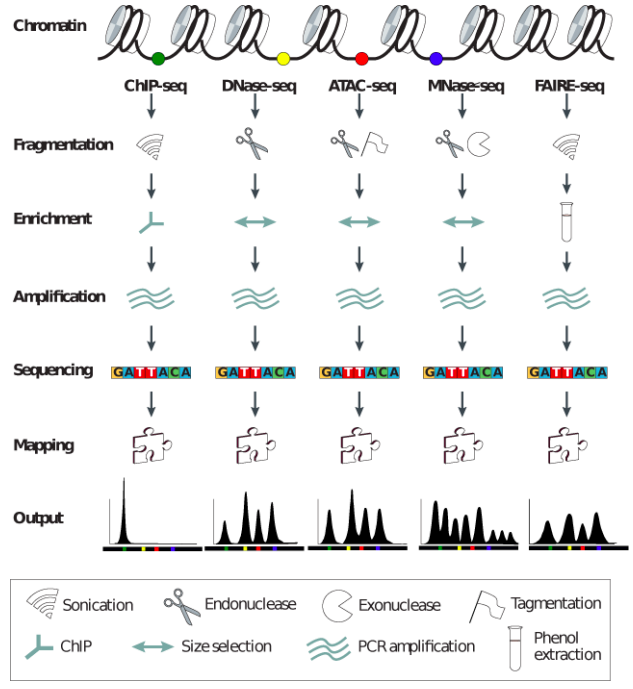
Meyer & Liu Nat. Rev. Gen. 2014
Data processing of Seqs:
Meyer & Liu Nat. Rev. Gen. 2014
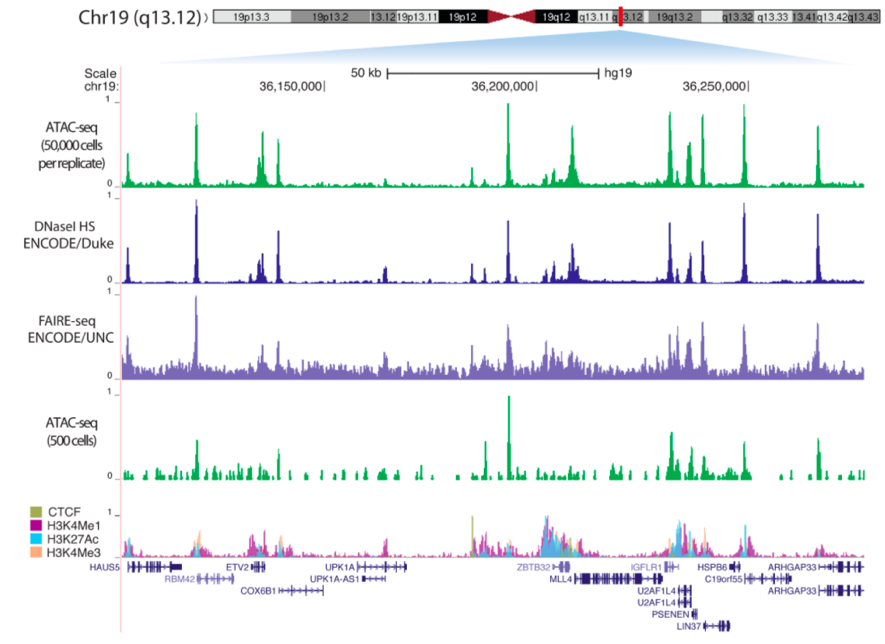
Examples of tracks:
Meyer & Liu Nat. Rev. Gen. 2014
In theory:

ATAC-Seq
Hsu et al. 2018
paired-end sequencing and mapping
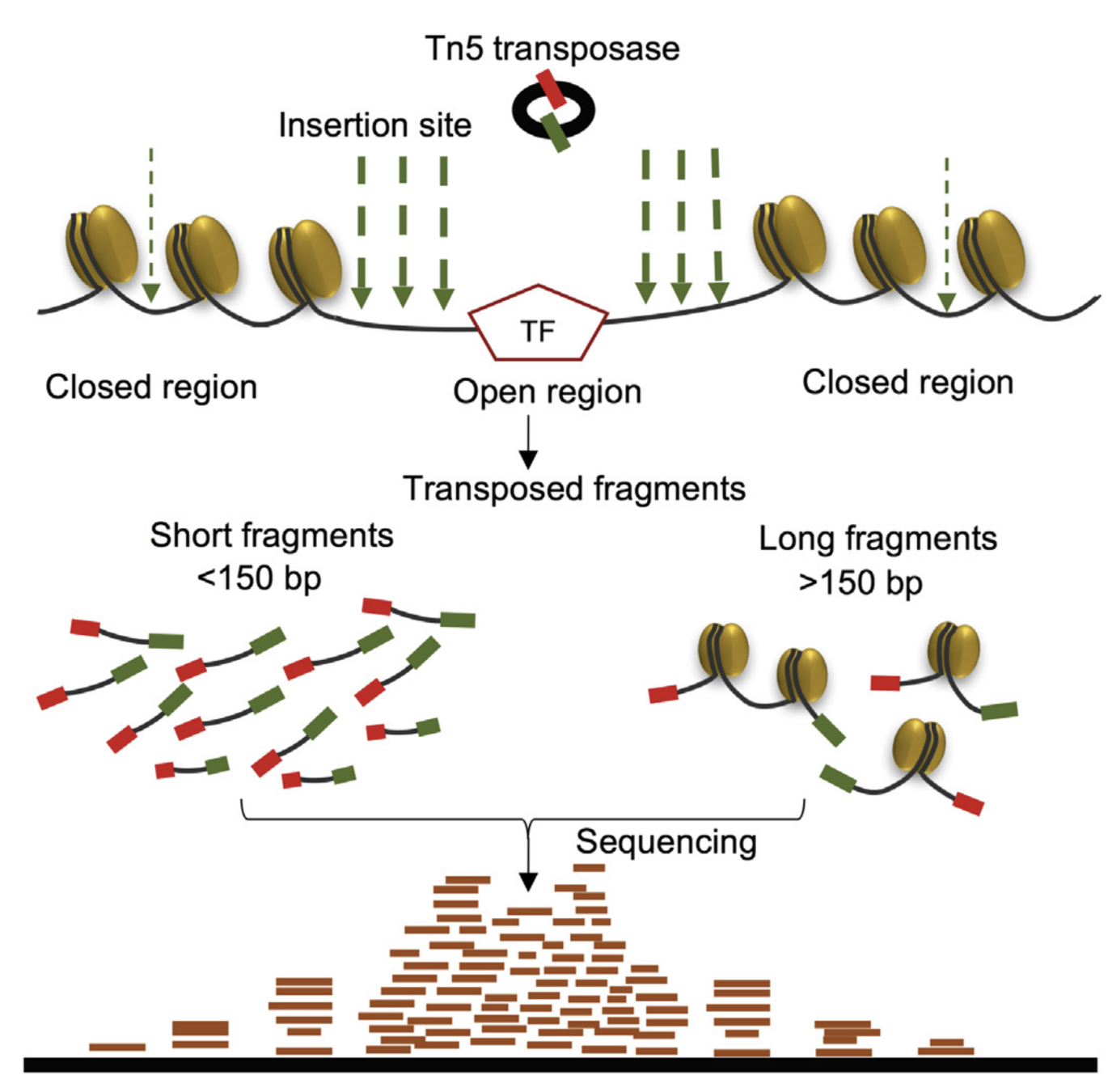
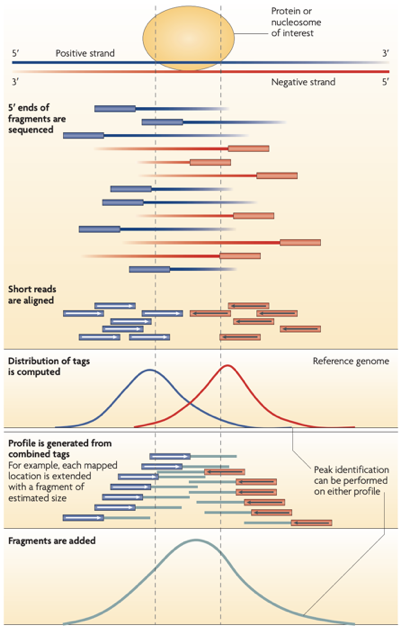
ATAC-Seq: insertion size distribution
Buenrostro et al., 2013
- Distance between forward and reverse mapping positions:

ATAC-Seq: insertion size distribution
Buenrostro et al., 2013
- Distance between forward and reverse mapping positions:
ATAC-Seq: insertion size distribution
Buenrostro et al., 2013
- Distance between forward and reverse mapping positions:
Nucleosome-free fraction
Nucleosome-bound fraction
ATAC-Seq around gene starts
Buenrostro et al., 2013
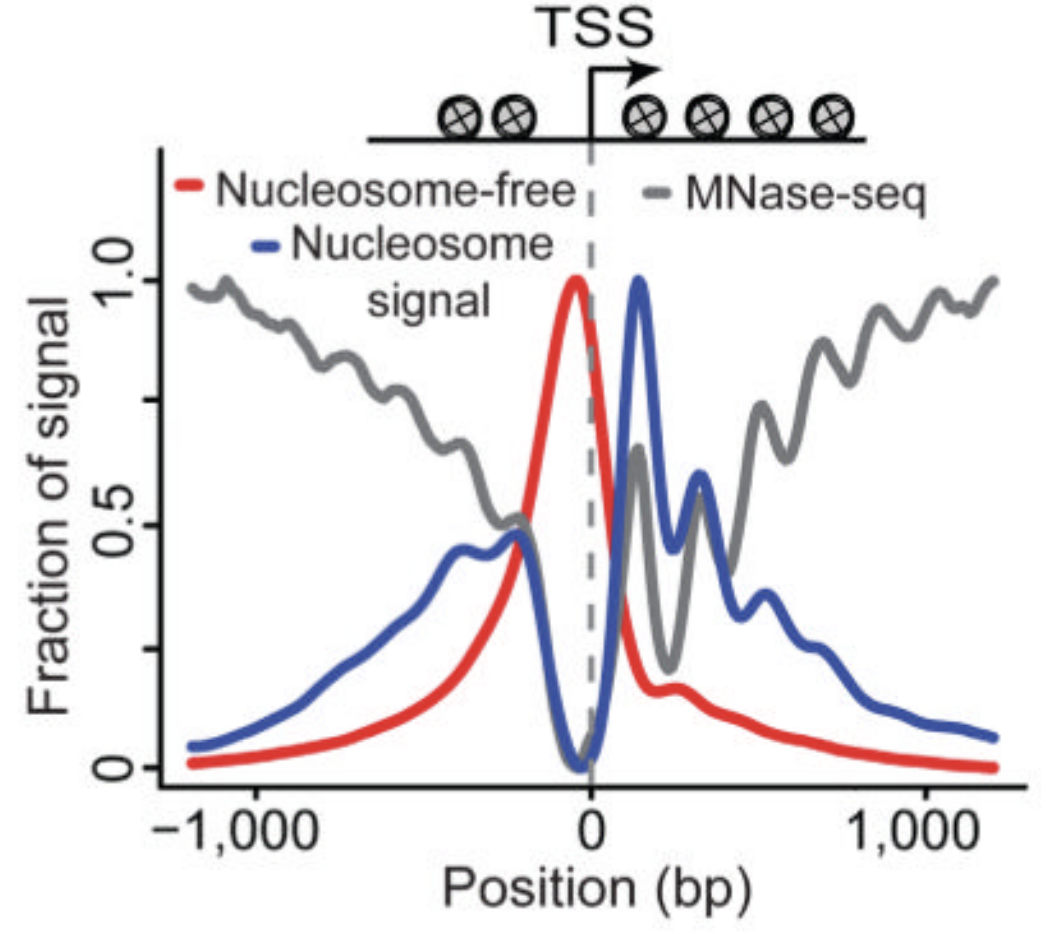
Nucleosome-free fraction
Nucleosome-bound fraction
- Average plot around
Transcription Start Sites (TSS):
ATAC-Seq around TF binding sites
Albanus et al., 2019
We can plot V-plot around positions of factors binding sites:
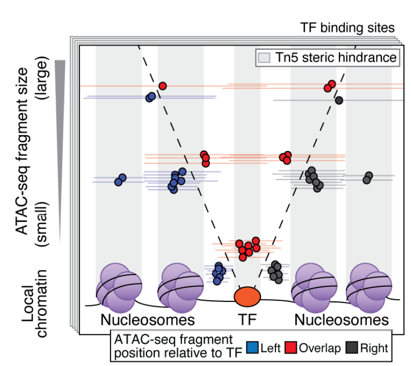
ATAC-Seq around TF binding sites
Albanus et al., 2019
ATAC-Seq around nucleosomes
Schep et al., 2015
We can also plot V-plot around positions of nucleosomes:
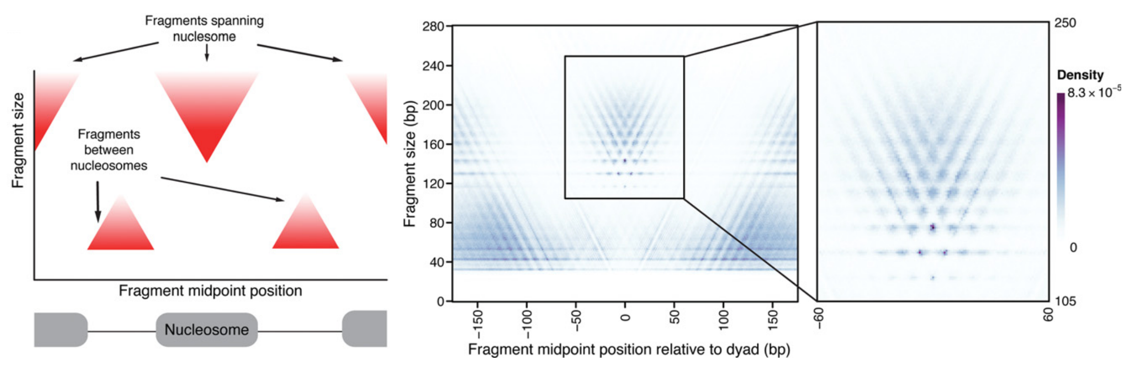
Chromatin openness (accessibility)
Hsu et al. 2018, Slattery et al. 2014
We can also do motifs search in open chromatin regions
Binding events and DNA motifs
Specific and non-specific binding:
Slattery et al. 2014
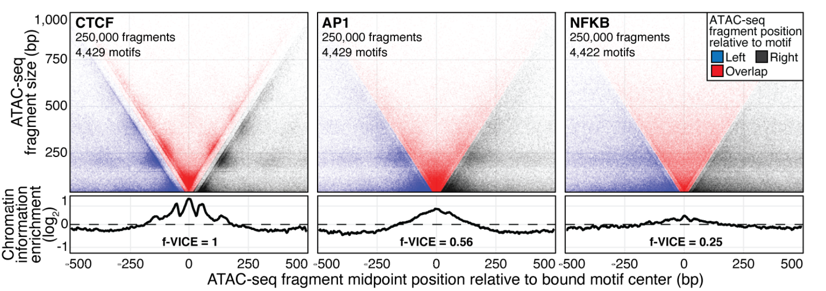
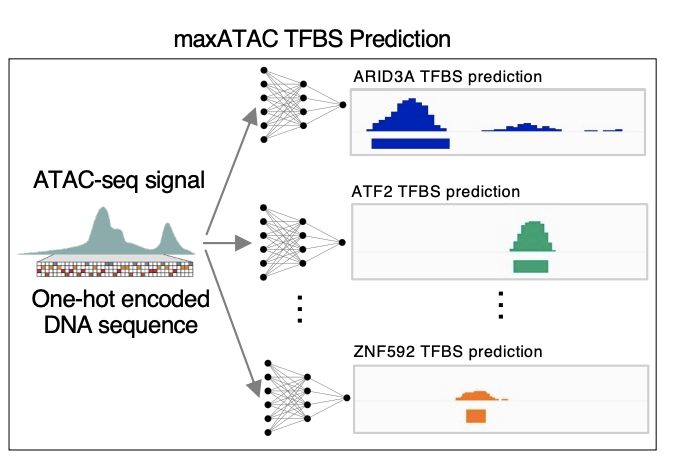
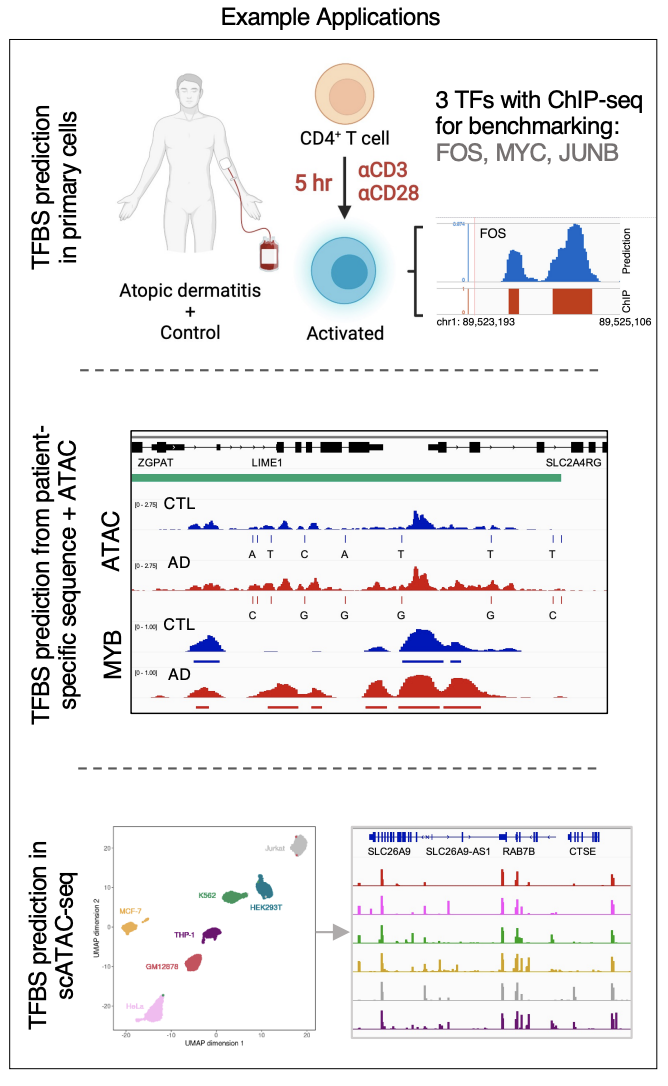
maxATAC: automated search of motifs in ATAC-Seq peaks
Cazares et al. 2022, bioRxiv preprint
High-resolution antibody-targeted chromatin profiling
Kaya-Okur and Henikoff et al. 2019 Efficient low-cost chromatin profiling with CUT&Tag
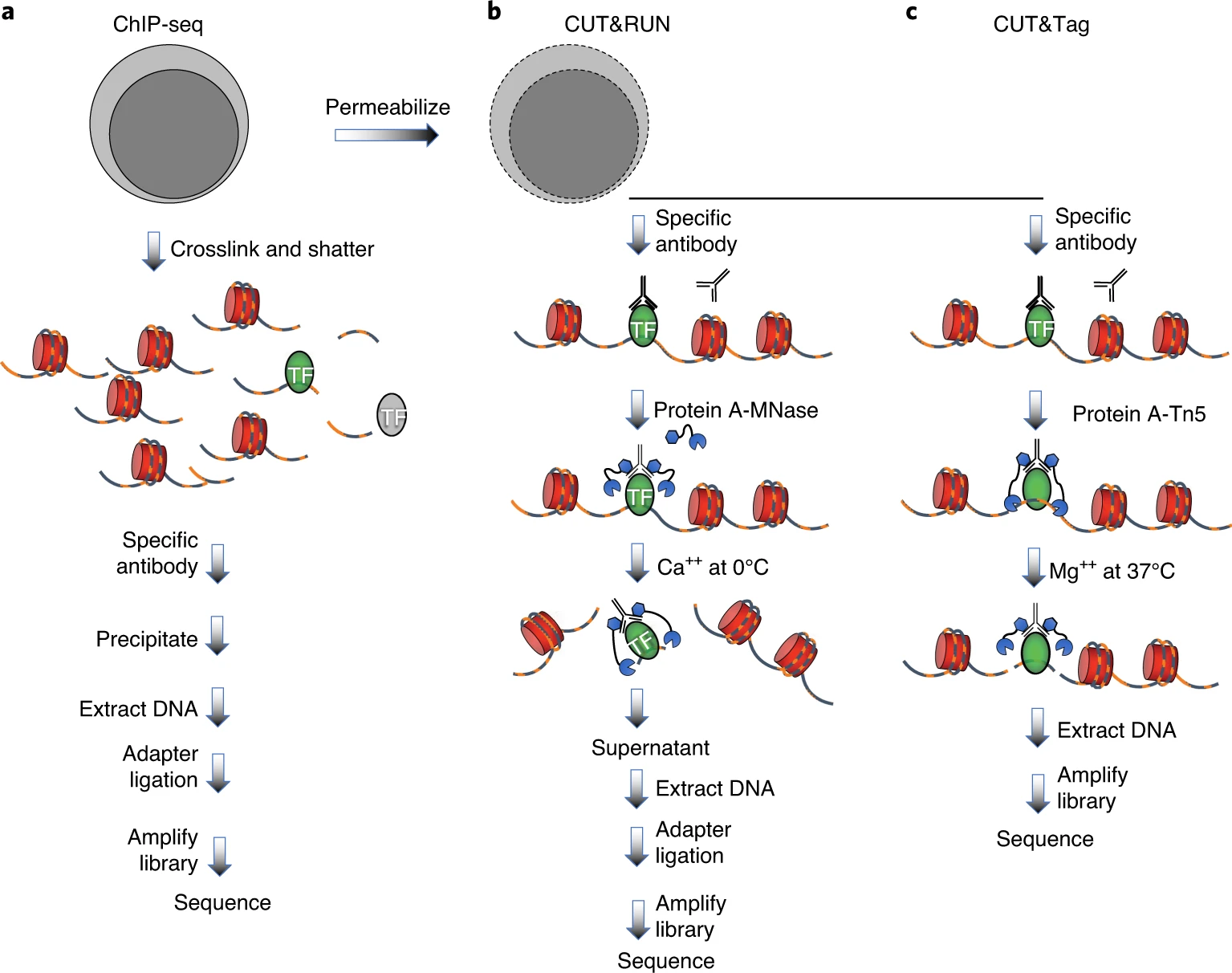
Cleavage Under Targets and Release Using Nuclease (CUT&Run)
Cleavage Under Targets & Tagmentation (CUT&Tag)
High-resolution antibody-targeted chromatin profiling
Kaya-Okur and Henikoff et al. 2019 Efficient low-cost chromatin profiling with CUT&Tag
Cleavage Under Targets and Release Using Nuclease (CUT&Run)
Cleavage Under Targets & Tagmentation (CUT&Tag)
Few slides on epigenetics data association
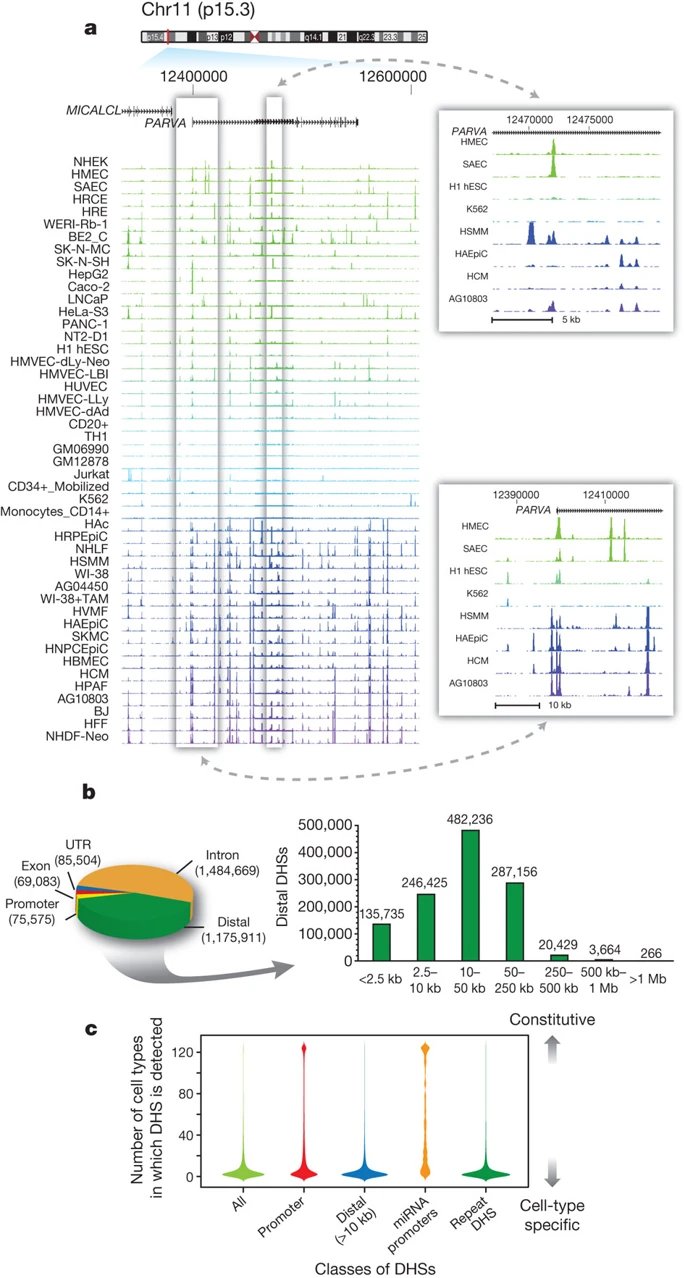
Genomics tracks association
DNase-Seq coverage for ∼350-kb region:
Thurman et al. Nature 2012
Different cell types
Clustering of samples
Cusanovich et al. Cell 2018
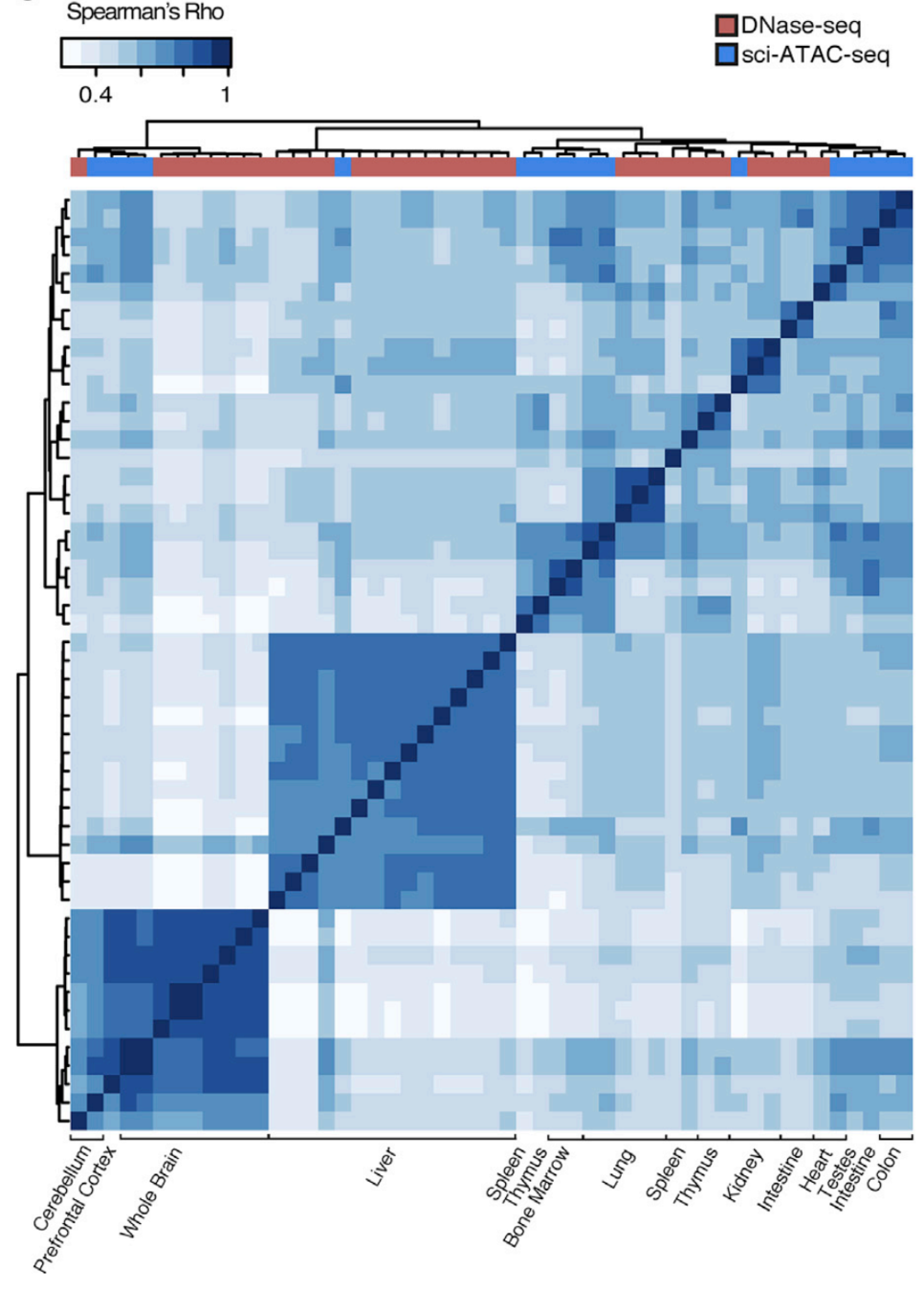
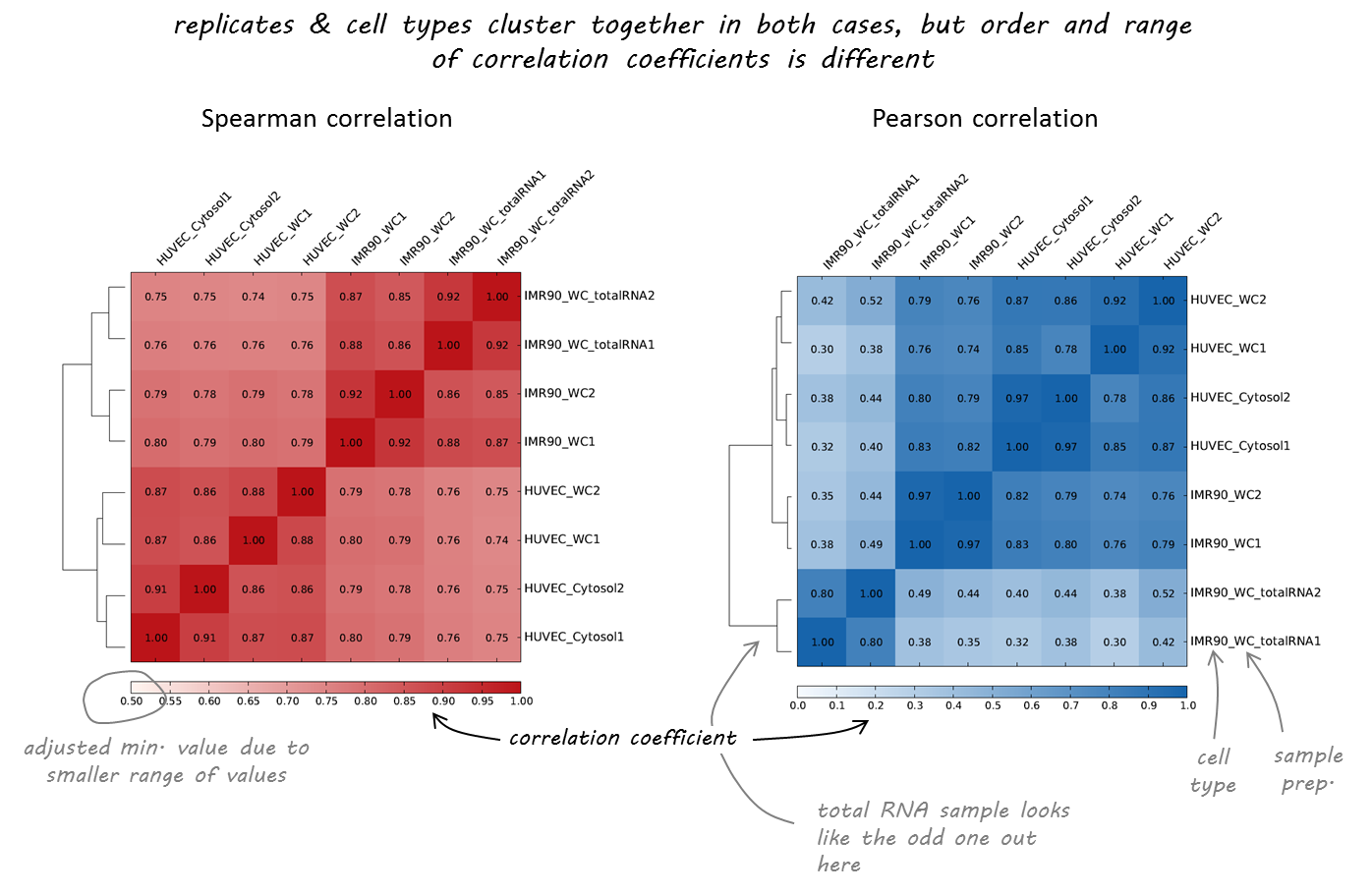
Let's check the similarity of pooled tissue samples with DNase-seq from known tissues. For that, let's plot bi-clustered heatmap of Spearman correlation coefficients:
Result of clustering,
the tree reflecting the similarity between samples
Clustering of samples
Cusanovich et al. Cell 2018
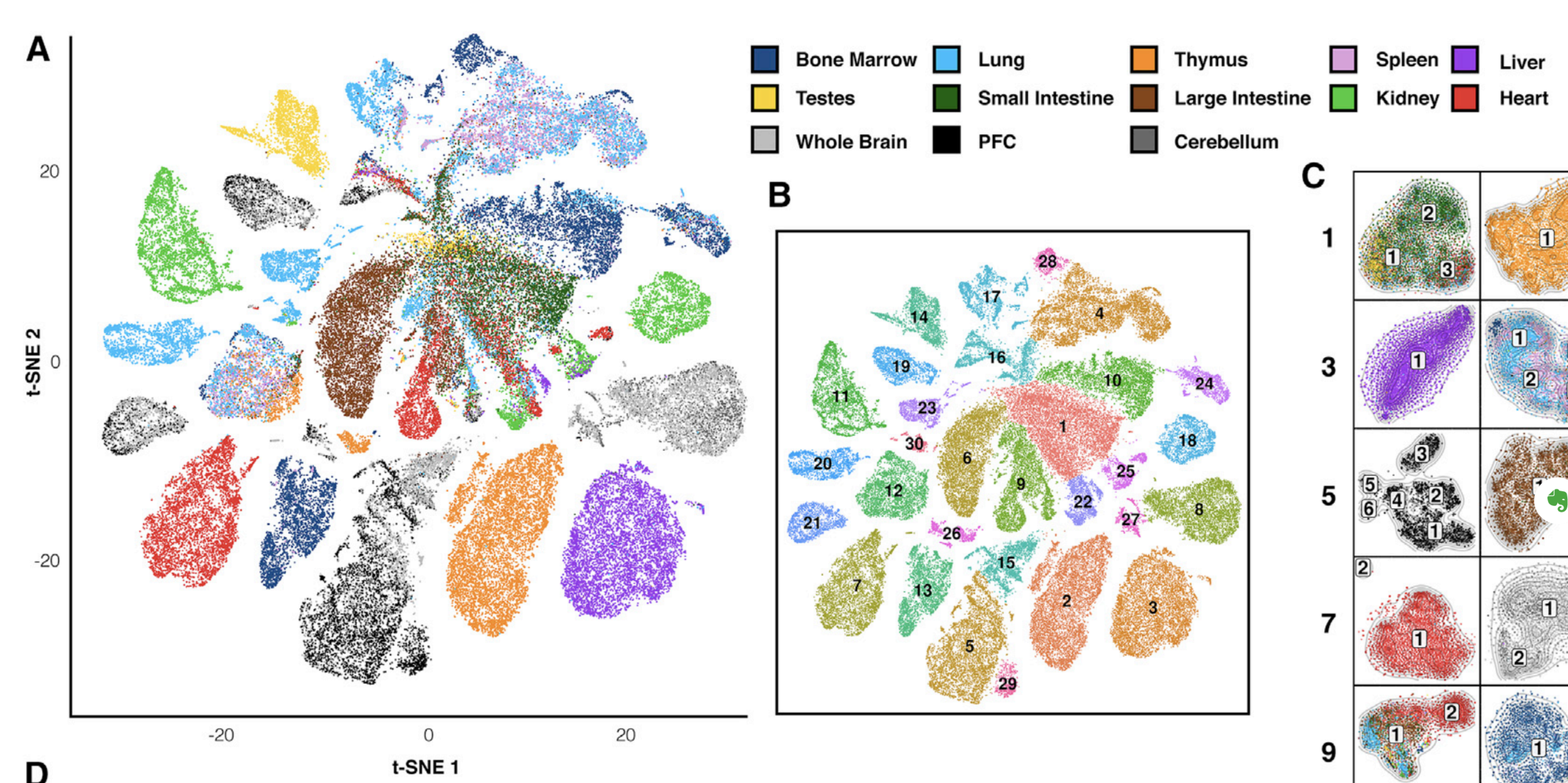
Finally, let's do dimensionality reduction, where each dot is a single sample.
Here is an example with t-SNE, although PCA (Principal Component Analysis) and MDS (Multidimensional scaling) are other popular techniques:
Practical Part: Samples Comparison
We will use deeptools commands multiBigwigSummary and plotCorrelation to compare the ChIP-Seq experiments. We will construct bi-clustered heatmaps of correlations:
Text
Stackup Plots (enrichment profiles):

Practice
Before we start the practice...
- Hometask made available on canvas:
- Find and follow the instructions for the seminar here
Before we start the practice...
- We will work in browser and with terminal. We also need to copy files from the remote server, so install appropriate software, if needed (your favourite terminal distributive,
Putty: https://www.putty.org/ or WinSCP: https://winscp.net/eng/download.php) - File formats are specified after the dot (e.g. .bam).
- Code is running in grey boxes with monospace font, try not to copy it blindly, because:
some file names are surrounded by brackets "${}". This indicates that it should be replaced by the name of your file. You may use the filenames that you like, but I highly recommend to stick to using the name of your factor and input instead.
echo "Bioinformatics has never been easier! Copy&paste wisely, or rm -rf ~/ will strike you one day."
Before we start the practice...
Review of some useful bash commands:
- List files in the current directory:
- Check the current position in the directory tree:
- Open text file for reading:
- Run command help, one of the options:
(some of them work for ones
but not the others)
- Copy file:
- Enter directory:
- Exit the directory:
- Create directory:
- File decompression:
lspwdless file.txtsome_command -hsome_command --helpman commandcp target_file destination_foldercd directory_name/cd ../ls -htsmkdirgzip -d file.gzunzip file.zipExpected result of the seminar
- Quizz on canvas (10 points in total)
- Extended deadline until 24th of March.
- Each task of the quizz is screenshot + essay answer.
- Mistakes on biology and theory are penalized.
- Feel free to ask the questions in the chat!
Additional slides
FYI
Get in touch with your data: step 1
1. Login to the server via terminal:
or use Putty on Windows.
(example instructions https://www.ssh.com/ssh/putty/windows/#sec-Configuration-options-and-saved-profiles)
2. Create the folder with your project:
3. Activate prepared environment:
(if you have problems with it, please, contact the TA)
4. Check your environment:
ssh username@servernamemkdir EpiPract1
cd EpiPract1export PATH="/home/a.galitsyna/conda/bin:$PATH"
conda activate chipseqls # Should list all the filesin the directory
pwd # Should result in your currentl location
conda list # Should list all the programs installed, if conda imported successfully.Get in touch with your data: step 2
- Find your datasets (here or in the table on the right):
- Copy to the local folder:
- Check the content and file sizes:
less /home/a.galitsyna/datamkdir ~/lesson6/
cp /home/a.galitsyna/data/reads/${your_files.fastq.gz} ~/lesson6/cd ~/lesson6
ls -hts Get in touch with your data: step 3
- Open FASTQ files, check the format
(see also: http://emea.support.illumina.com/bulletins/2016/04/fastq-files-explained.html)
- Check FASTQ files, what is the number of rows vs the number of entries?
- Run fastqc (a tool for FASTQ quality control, or QC)
gzip -d <file.fastq.gz> # This will result in <file.fastq> unzipped file
less <file.fastq>wc -l <file.fastq>fastqc <file.fastq>NGS data typical workflow example
Let's review the NGS data processing workflow:
- Quality control: fastqc on initial FASTQ
- Index build: bowtie2-build on initial chromosomes files
- Read mapping: FASTQ → SAM/BAM
- Filtering of mapped reads: samtools on SAM from p.3
- Conversion to BED: bedtools on SAM from p.4
FASTQC report example
- Takes FASTQ as input:
- Output format: html and plain text
fastqc <input.fastq>ls
# input.fastq input_fastqc/
cd input_fastqc/
ls
# fastqc_data.txt fastqc_report.html Icons Images summary.txtDon't run this code, it's FYI
FASTQC report example
- Example html output:
(see https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ for more examples)
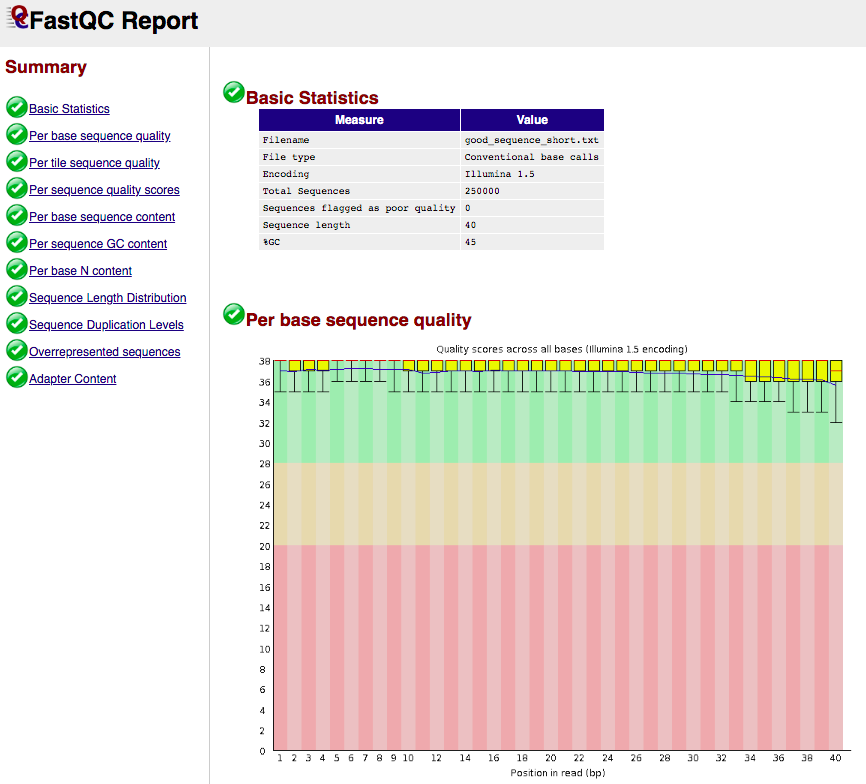
Genome index build example
(Manual: http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml)
- Required for fast search of query sequence during mapping
- Takes fasta with reference as input (comma-separated list) and prefix name.
- Example run and resulting database:
bowtie2-build -h
bowtie2-build chr1.fa,chr2.fa,chr3.fa,chr4.fa hg38
# Check the results:
ls -hs
# 938M hg38.1.bt2 701M hg38.2.bt2 12K hg38.3.bt2 701M hg38.4.bt2 3.0G hg38.fa 938M hg38.rev.1.bt2 701M hg38.rev.2.bt2
bowtie2-inspect -n hg38Don't run this code, it's FYI
Read mapping example
- Takes query FASTQ and indexed database as input.
- Output is in SAM or BAM format
(SAM format specification: https://samtools.github.io/hts-specs/SAMv1.pdf) - SAM is a TSV file with header, BAM is its compressed binary version. The columns are:
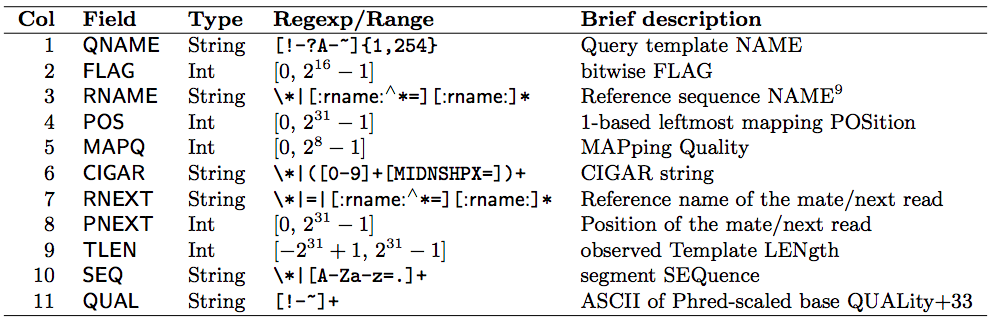
Read mapping example
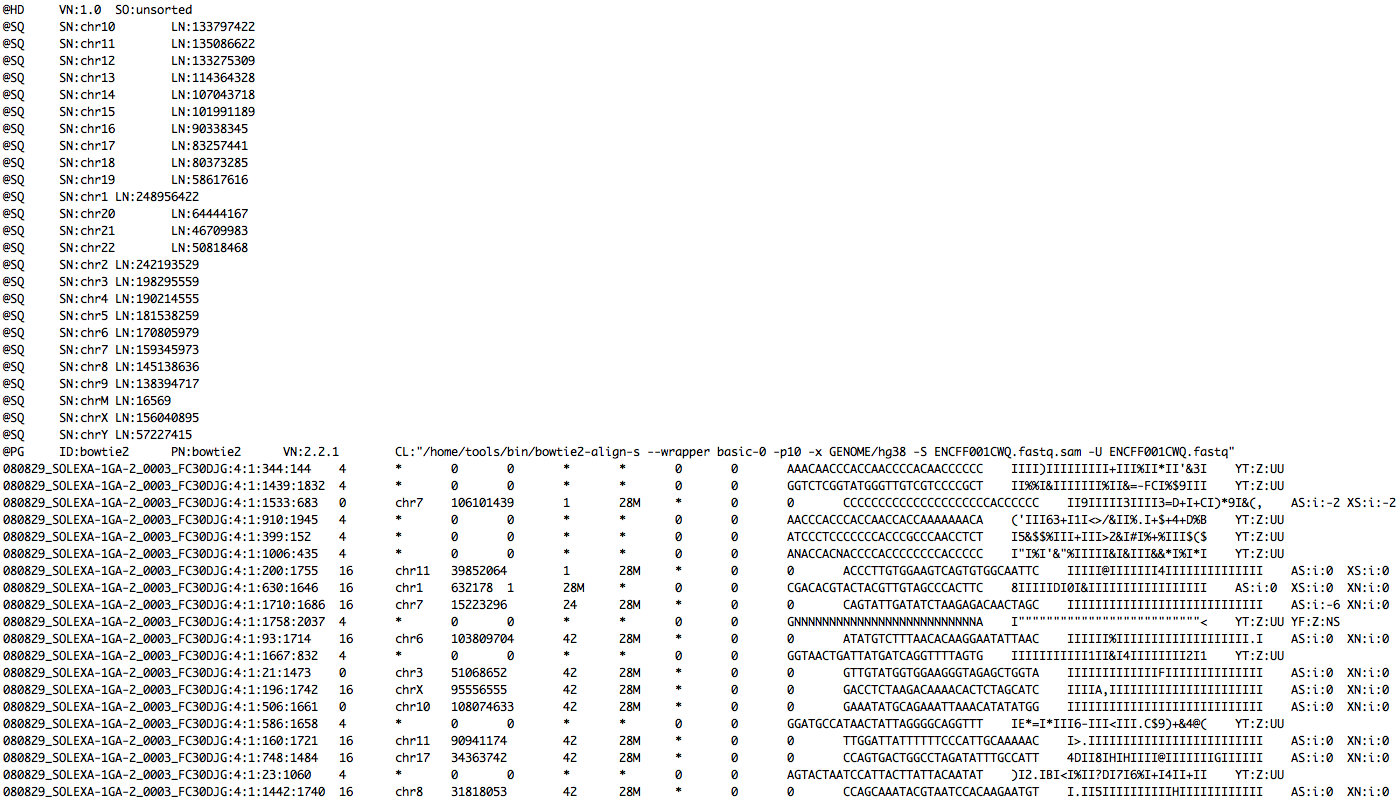
FASTQ
+ indexed database

-> SAM file
Samtools example
(Manual: http://www.htslib.org/doc/samtools.html)
- A toolkit designed for SAM/BAM manipulations
- View:
- Statistics assessment:
- Format conversion:
- Filtering:
samtools view file.samsamtools view -F 4 input.sam
samtools stats file.samsamtools view -b file.sam > file.bamBedtools example
(Manual: https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html
BED format specification: https://genome.ucsc.edu/FAQ/FAQformat.html)
- A toolkit for conversion and manipulations with BED:
- A powerful tool for genomic arithmetics:
(Tutorial: https://bedtools.readthedocs.io/en/latest/)
bedtools bamtobed -i file.bam > file.bed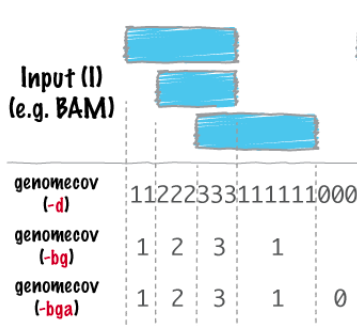
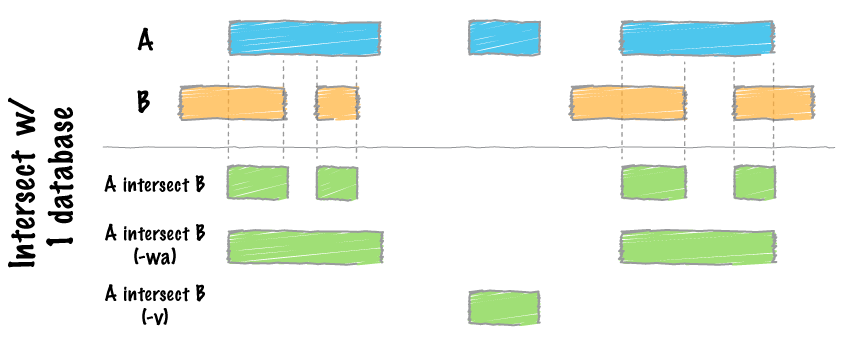
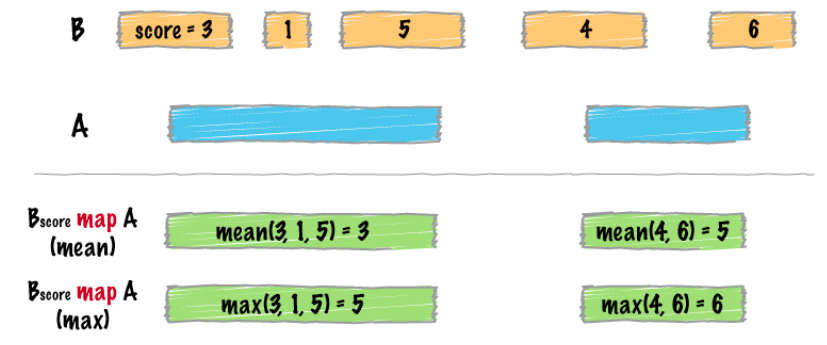
map
intersect
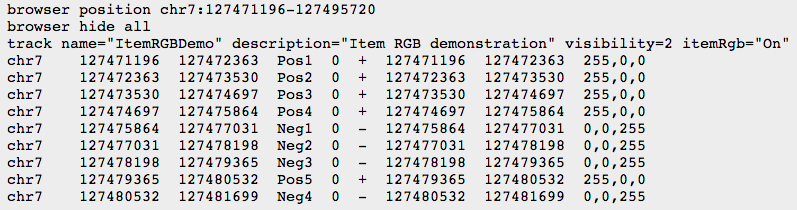
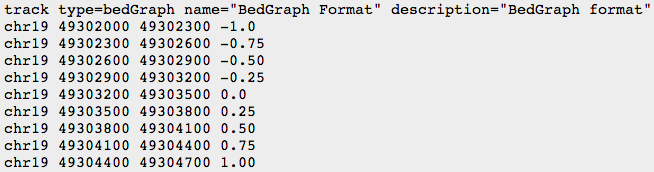
Other formats
- BED
- BEDGRAPH
- BIGWIG: binary compressed format, created with UCSC tools (see below)
UCSC genome browser & UCSC tools
- UCSC maintains genome browser and has a set of tools: https://genome-euro.ucsc.edu/

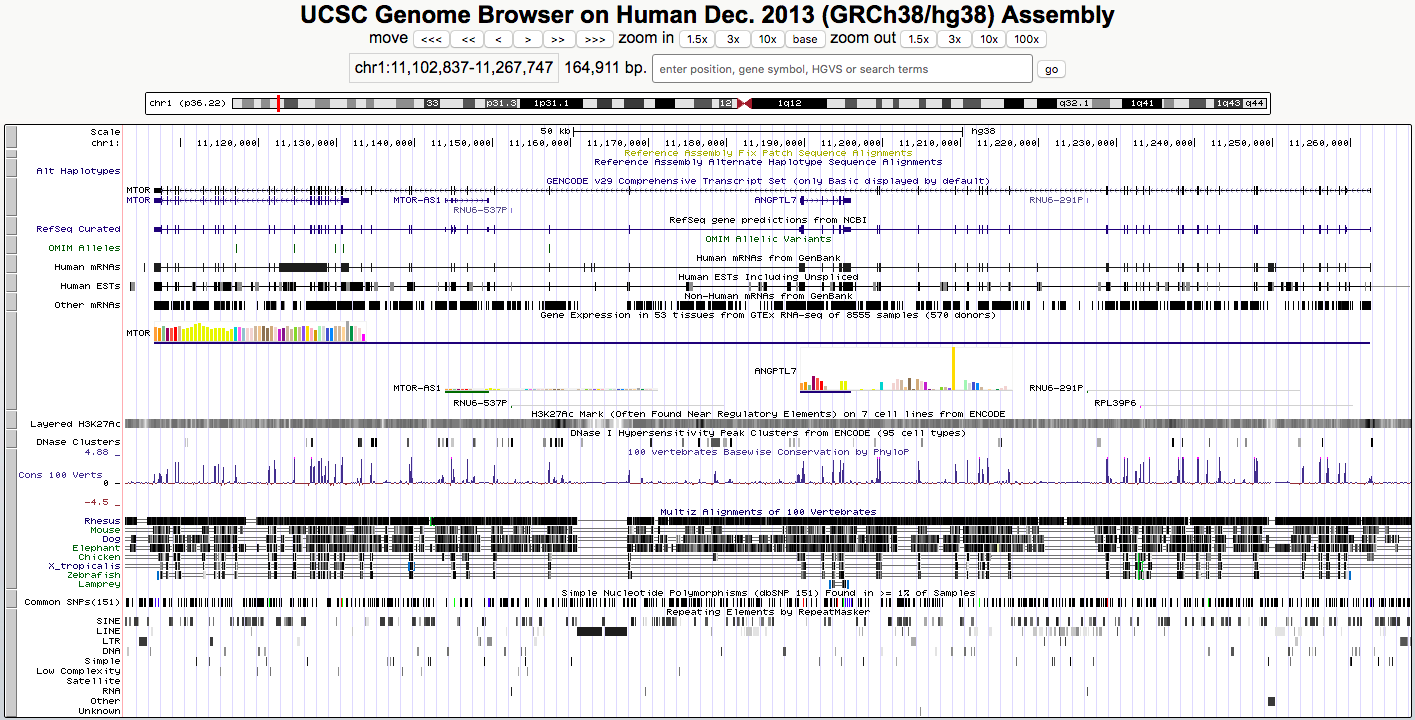
UCSC tools example
Full set of compiled tools: http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
- We will need file conversion, check the documentation:
bedGraphToBigWig -hNext step: index build and mapping
These steps are time-consuming, but we have to complete them:
- Build genome index with name prefix hg38_chr5-6_index:
- Run mapping of your FASTQ file for the index hg38_chr5-6_index:
cd GENOME/
bowtie2-build dm6.fabowtie2-build -hbowtie2 -hbowtie2 --very-sensitive -x ./GENOME/dm6 <file.fastq> -S <file.sam>less <file.sam>ls
cd ../Don't run this code, it's FYI
Filtering
- Filter only uniquely mapped reads with high alignment score, sort resulting file:
samtools view -h -F 4 <file.sam> | lesssamtools --helpsamtools view -h -q 10 <file.sam> > <file_filtered.sam>samtools view -b -q 10 <file.sam> > <file_filtered.bam>
samtools sort <file_filtered.bam> > <file_filtered.sorted.bam>Don't run this code, it's FYI
File format conversion
- Read the documentation of bedtools:
- Convert BAM to BEDGRAPH genome coverage:
- Create files with genomic windows (1Kbp bin size, don't forget to use your chromosome!). Convert BAM to BEDGRAPH coverage of windows:
bedtools makewindows -g GENOME/chrom_sizes.tsv -w 1000 > hg38_windows.1000.bed
grep "chr5" hg38_windows.1000.bed > chr5_windows.1000.bed
bedtools genomecov -ibam <file_filtered.sorted.bam> -bga -split > <file.genomecov.bedgraph>bedtools genomecov -h
bedtools makewindows -h
bedtools coverage -h
bedtools coverage -counts -a chr5_windows.1000.bed -b <file_filtered.sorted.bam> > <file.binned.bedgraph>- Check the resulting files, you should see genome coverage
files (*.genomecov.*) and binned datasets (*.binned.*):
ls
less <file.genomecov.bedgraph>
less <file.binned.bedgraph>Don't run this code, it's FYI
BEDGRAPH tracks visualization
We will use UCSC browser for bedgraph files visualization.
- Download both files file.genomecov.bedgraph and file.binned.bedgraph to your local computer.
- Manually add the header to your file:
(see UCSC recommendations: https://genome.ucsc.edu/goldenPath/help/bedgraph.html)
track type=bedGraph visibility=full
- Go to UCSC genome browser, hg38 genome:
https://genome-euro.ucsc.edu/cgi-bin/hgTracks?db=hg38
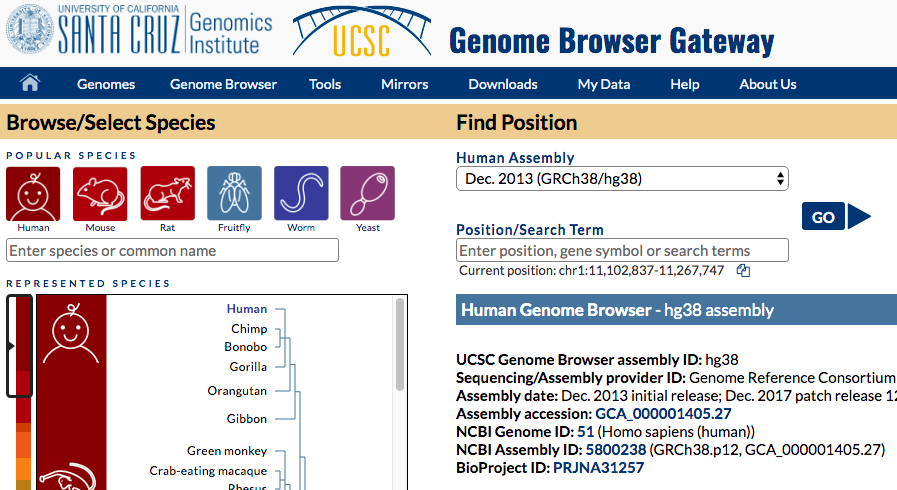
UCSC genome browser
- Find "add custom tracks button".
- Check the upload form:
- Upload your modified BEDGRAPH files (with headers) to UCSC ("Choose file" -> "Submit").
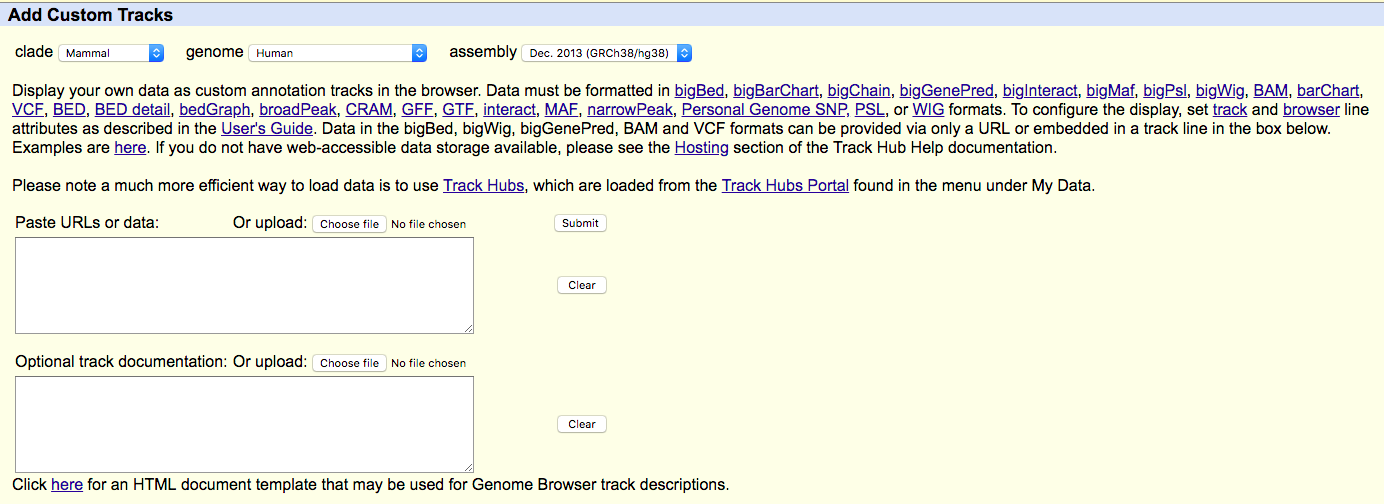
Experiments comparison
- Convert BEDGRAPH to BIGWIG:
- Rename your final file (substitute YOUR_CELL_LINE and CHROMOSOME):
- Find three of your colleagues with the same chromosome as you. Exchange your BIGWIG files and upload them to the server. Thus you should collect at least 4 BIGWIG files with recognisable names (cell line and chromosome should be present in the file names).
mv <file.genomecov.bw> <YOUR_CELL_LINE-CHROMOSOME.bw>bedGraphToBigWig <file.genomecov.bedgraph> ./GENOME/chrom_sizes.tsv <file.genomecov.bw>Samples comparison
Task 1 (1 point): Provide the final set of commands that you've used and briefly justify the parameters.
Task 2 (1 point): Report the final scatteplot for 100 Kb. Describe your observations.
Task 3 (*1 point): Repeat the procedure for 10 Kb and 1 Kb and report the changes in observations. At what resolution you might observe the differential DNase I signal?
- Collect at least 4 DNase I results for your chromosome in bigWig format. One of them should be yours, the others you may take from /home/shared/EpiPract1/. you may use conda environment "hic" (see instructions on activation can be found in EpiPract3).
- Use multiBigwigSummary to create a summary file. Use bins mode, bin size 100 Kb:
- Plot correlations obtained from your file as a scatterplot:
- Try to adjust the parameters of the plot to improve the analysis. Try plotting in log-scale (--log1p). If you get the warning about the outliers, try to fix it: use --removeOutliers and --corMethod spearman. Which method worked?
- * Repeat the procedure for 10 Kb and 1 Kb. You may want to create another intermediary summary file for that. Inspect the changes.
multiBigwigSummary bins --binSize 100000 -b ${file1.bw} ${file2.bw} ${file3.bw} ${file4.bw} -o ${summary.npz}plotCorrelation -in ${summary.npz} --corMethod pearson --skipZeros --whatToPlot scatterplot -o ${scatterplot.png}Experiments comparison
6. Plot correlations for 100 Kb as a heatmap. Use the parameters that you found to be representative for the scatterplots. For example:
7. Inspect and describe your observations. Adjust the visualization, if needed.
plotCorrelation -in ${summary.npz} --corMethod pearson --skipZeros --whatToPlot heatmap --colorMap RdYlBu --plotNumbers -o ${heatmap.png}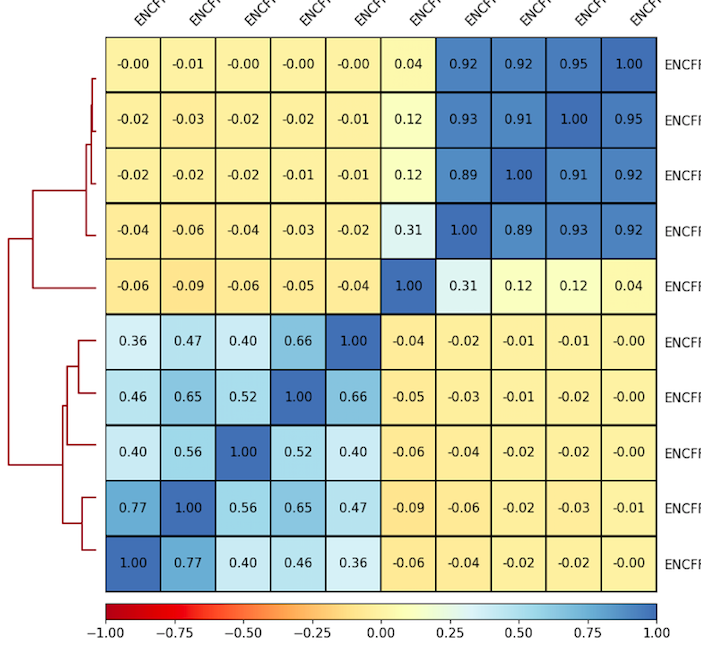
Result of clustering
Correlation coefficient
Labels are not representative
(correct if needed)
Practical Part: Enrichment Profiles
We will:
- Call TADs in S2 cell line (Wang et al. 2017),
- Plot the enrichment profiles for multiple factors.
We'll need conda environment "hic" (see instructions on activation can be found in EpiPract3).