Практическая работа с данными Hi-C
Александра Галицына
Цикл лекций о структуре хроматина
2 декабря 2021




Квиз №1 по структурной организации хроматина
Ссылка на форму:
время проведения:
11:10 до 11:30
(строгий дедлайн)
Пространственная организация хроматина
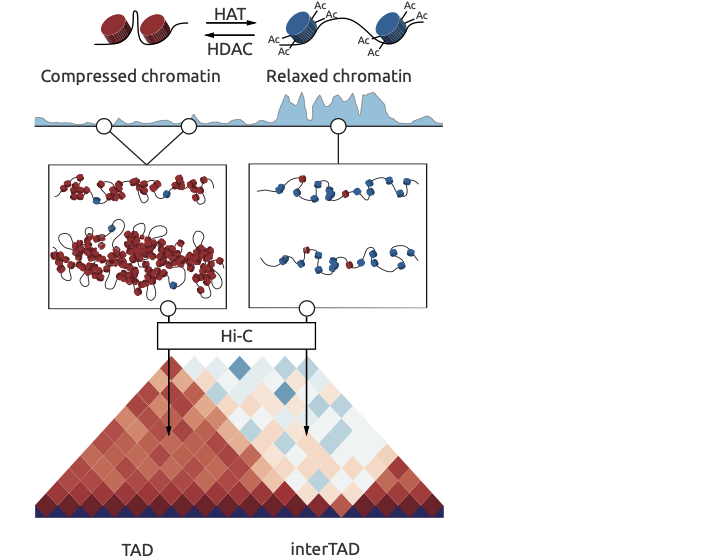
Ulianov et al. Genome Biology 2016

Модель выпетливания
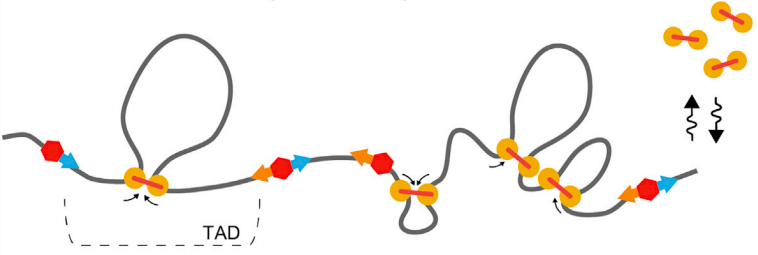
Fudenberg et al. 2016 Cell Reports

когезин - выпетливающий фактор
CTCF - граничный элемент
Подходы к изучению структуры хроматина
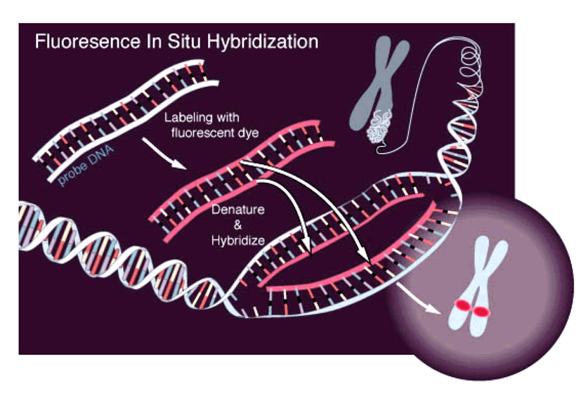
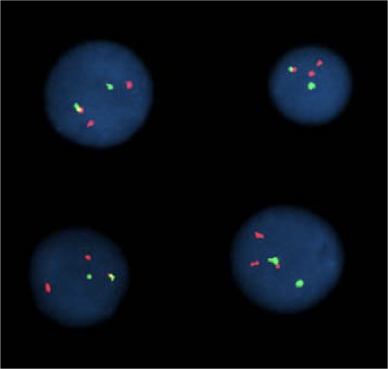
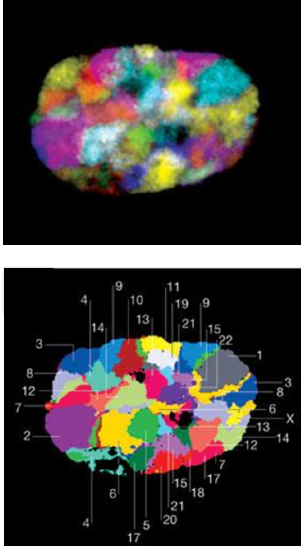
Микроскопия:
Микроскопия с флуоресцентными маркерами (FISH):
FISH на двух метках:
FISH для полных хромосом:
Chromosomes conformation capture (3C)
3C: Dekker et al. Science 2002
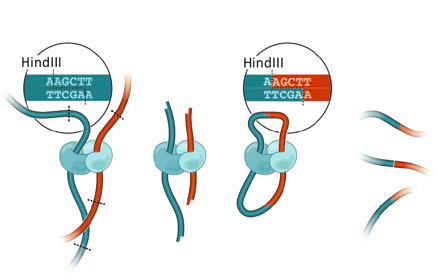
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Библиотека ДНК-ДНК контактов
Фиксация конформации хромосом
Hi-C (high-throughput CCC)
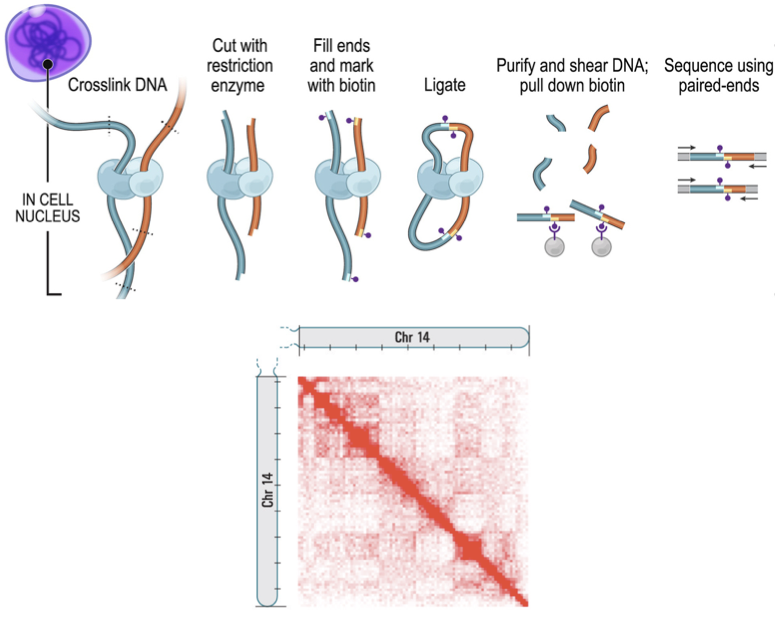
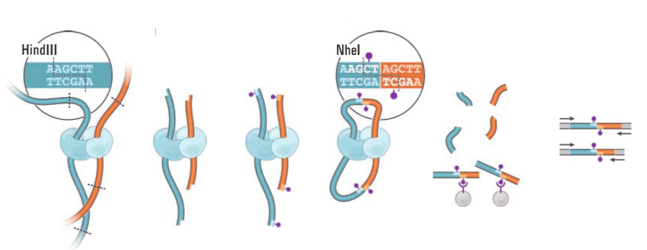
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Секвенирование
Картирование
Лигирование
ВЫСОКОпроизводительная фиксация конформации хромосом
Разнообразие методов фиксации конформации
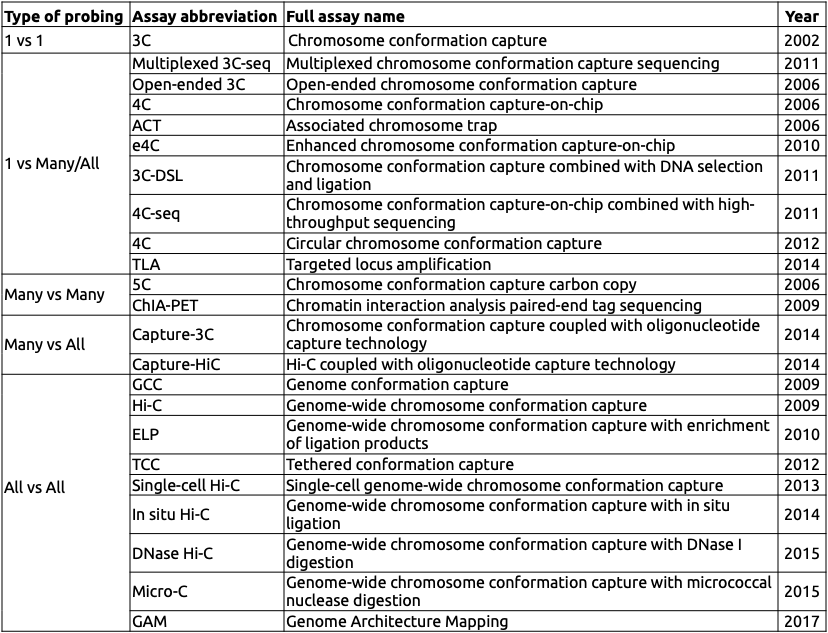
Adopted from Schmitt Nature Reviews 2016
Принципы организации структуры хроматина
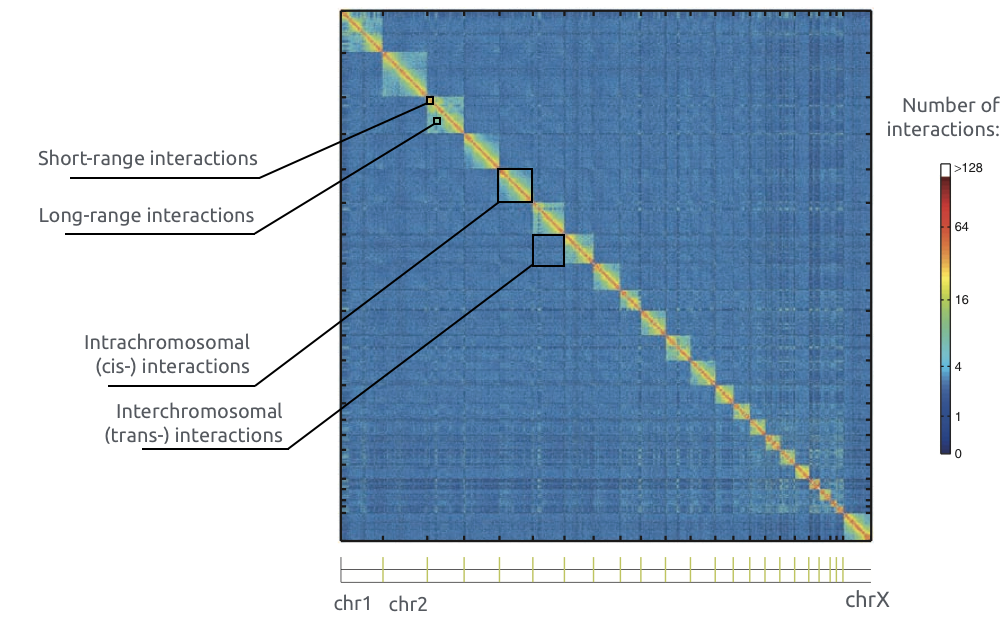
Adopted from Imakaev et al. Nature Methods 2012
Общий план обработки Hi-C
0. Контроль качества данных NGS
1. Картирование ридов:
-> получение файла с картированиями (.sam)
2. Конвертация файла с картированиями (parse):
-> получение файла с парами контактов (.pairs)
- дедупликация
3. Получение карты геномных контактов (.cool):
- бинирование
- нормализация и корректировка
5. Контроль качества Hi-C
6. Поиск особенностей (фич) карт Hi-C
<- в следующий раз
Практика:
"Зоопарк"
в хроматиновой лаборатории
Задание
Установка: вы работаете в биоинформатической лаборатории по исследованию эволюционных процессов в хроматине. Вам даны:
- данные Hi-C для некоторого организма,
-
база данных о геномах с аннотацией видов
Ваши задачи:
1) определить организм, для которого выполнен Hi-C;
2) провести процедуру обработки данных Hi-C;
3) вынести вердикт: хорошо ли сработал протокол Hi-C и секвенирование
4) провести интерпретацию результата и предложить гипотезы об особенностях укладки хроматина организма
Форма для ответов:
https://forms.gle/dqBa4ENySyCywD2T6
Максимально 10 баллов за ответ. Ошибки можно компенсировать за счет последнего задания на биологическую интерпретацию результатов.
Дедлайн: 9 декабря в 11:10
(будем разбирать правильные ответы!)
Кластер
1. Логин через терминал:
или Putty для Windows.
(инструкции: https://www.ssh.com/ssh/putty/windows/#sec-Configuration-options-and-saved-profiles)
2. Активируйте удобный bash-терминал:
3. Создайте директорию проекта:
4. Настройка рабочей среды и ее проверка:
$ ssh -p5222 username@92.242.58.92 или ssh -p32222 username@92.242.58.92$ mkdir practice_chromatin
$ cd practice_chromatin$ /usr/share/data-minor-bioinf/chromatin/anaconda/condabin/conda init
$ bash
$ conda activate chromatin
$ cooltools --version$ bashДанные
- База данных геномов:
- Аннотация базы данных геномов:
https://docs.google.com/spreadsheets/d/1lhHjgq2HT83RCr_EBU9IyD2iQ7paUIHG-5Qh95pLd28/edit?usp=sharing
3. База данных секвенирования:
4. Ваш уникальный идентификатор данных:
https://docs.google.com/spreadsheets/d/1rJA3Cz1itIv8Z_cBjMaBK1vpKnYe7kOKzGrsrCmrw9E/edit?usp=sharing
ls /mnt/storage/chromatin/genomes/В папках содержится информация:
1. Прочтения секвенирования, первые и вторые из пар:
/mnt/storage/chromatin/fastq/<YOUR CODE>_R1.fastq
/mnt/storage/chromatin/fastq/<YOUR CODE>_R2. fastq
2. Файл с размерами хромосом данного организма:
/mnt/storage/chromatin/genomes/<YOUR GENOME ID>/<YOUR GENOME ID>.fa.sizes
3. Индекированный геном организма:
/mnt/storage/chromatin/genomes/<YOUR GENOME ID>/index/bwa/<YOUR GENOME ID>.fa*
ls /mnt/storage/chromatin/fastq/Определение вида
- Откройте Ваш файл с прочтениями Hi-C (прямые или обратные прочтения) для просмотра
- Возьмите несколько последовательностей ДНК из середины файла
- Воспользуйтесь nucleotide blast для того, чтобы найти схожие последовательности в нуклеотидных базах данных:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
Не доверяйте результатам только по одному риду. Попробуйте несколько штук из разных фрагментов файлов, чтобы избежать попадания в контаминированные риды (например, симбионтами, бакретиями в лабе или ДНК лаборанта-экспериментатора...)
less /mnt/storage/chromatin/fastq/1_R1.fastq
Несколько слов о сборке генома
- Найдите организм, который ближе всего к находке в BLAST из списка геномов. Откройте файл с хромосомами на кластере.
Перед Вами может оказаться:
- бактерия с одной хромосомой
- эукариот с геномом, собранным до хромосом
- эукариот с геномом, собранным до контигов
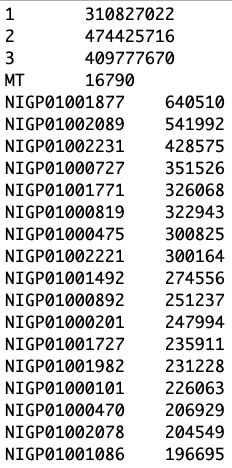
less /mnt/storage/chromatin/genomes/AaegL5/AaegL5.fa.sizesНесколько слов о сборке генома
Контроль качества данных NGS
$ fastqc ${yourfile.fastq} -o ./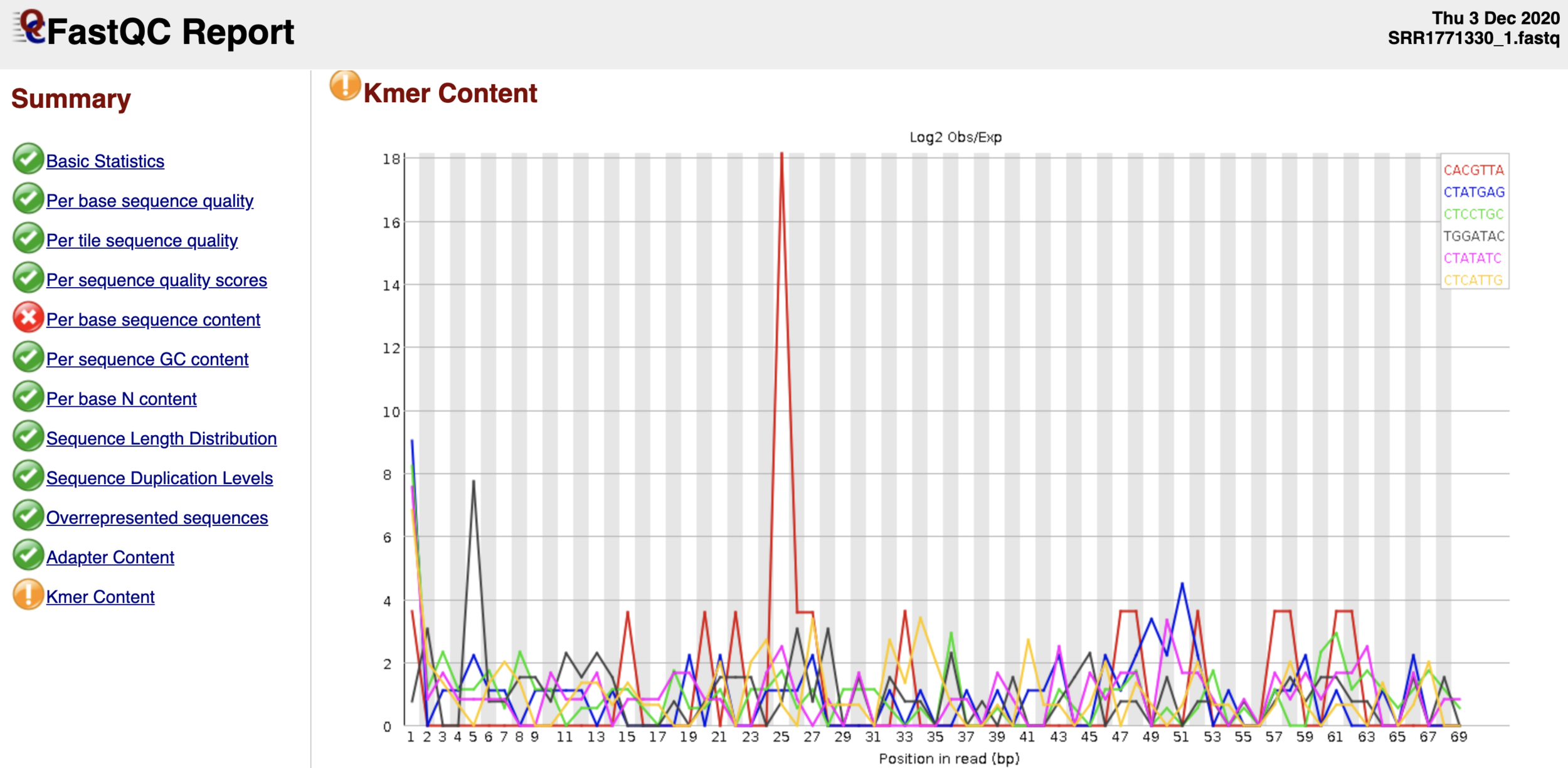
- Запустим fastqc и посмотрим на html-репорт (нужно скачать):
(*) после знака $ в фигурных скобках указано имя файла, которое нужно заменить на свое
Картирование данных
Картирование, переводит
fastq (файл с ридами) в sam (файл с выравниваниями):
- ! Может работать >40 минут для некоторых геномов
- Проверяйте загруженность кластера в параллельном терминале:
- Используйте запуск в фоновом режиме, если есть риск отключения кластера (с осторожностью):
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam}htop
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam} &Особенности данных Hi-C
! Секвенирование парноконцевое
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Лигирование
Типы пар прочтений в Hi-C
Imakaev et al. Nature Methods 2012
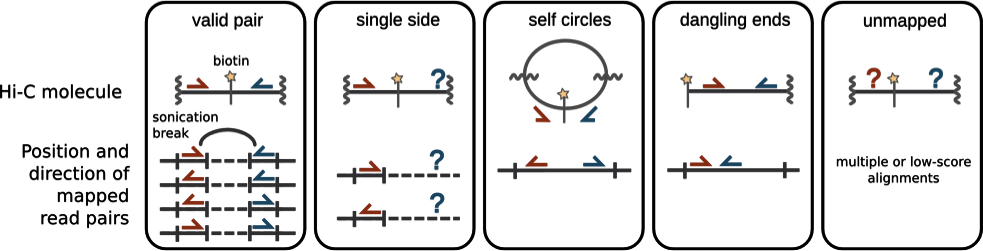
Структура пары прочтений Hi-C

хороший сценарий картирования
риды (.fastq)
выравнивания (.sam/.bam)
контакты (.pairs)
тип контакта (техническая информация pairtools)
Структура пары прочтений Hi-C

пара ридов не картировалась
Структура пары прочтений Hi-C
множественные картирования

Структура пары прочтений Hi-C
химерные прочтения - один рид перекрывает несколько фрагментов генома
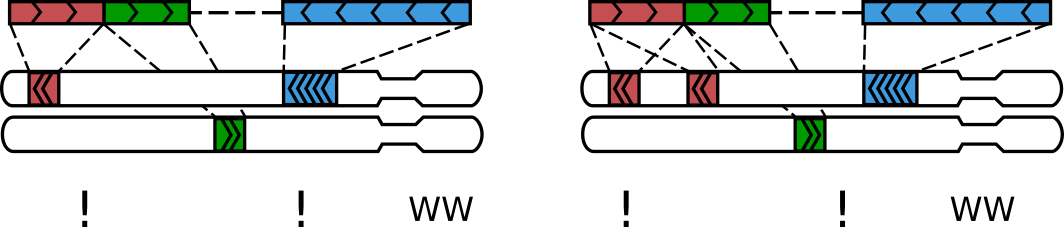
Итеративное картирование
Adopted from Lajoie et al., The Hitchhiker's guide to Hi-C analysis: Practical guidelines.
Methods 2015
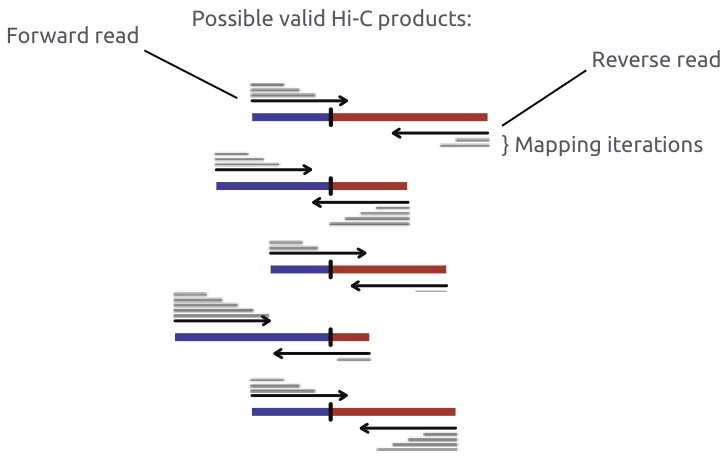
Один из первых методов решения проблемы химерных ридов (сейчас остался в истории):
Структура пары прочтений Hi-C
Сквозные прочтения
(readthrough)
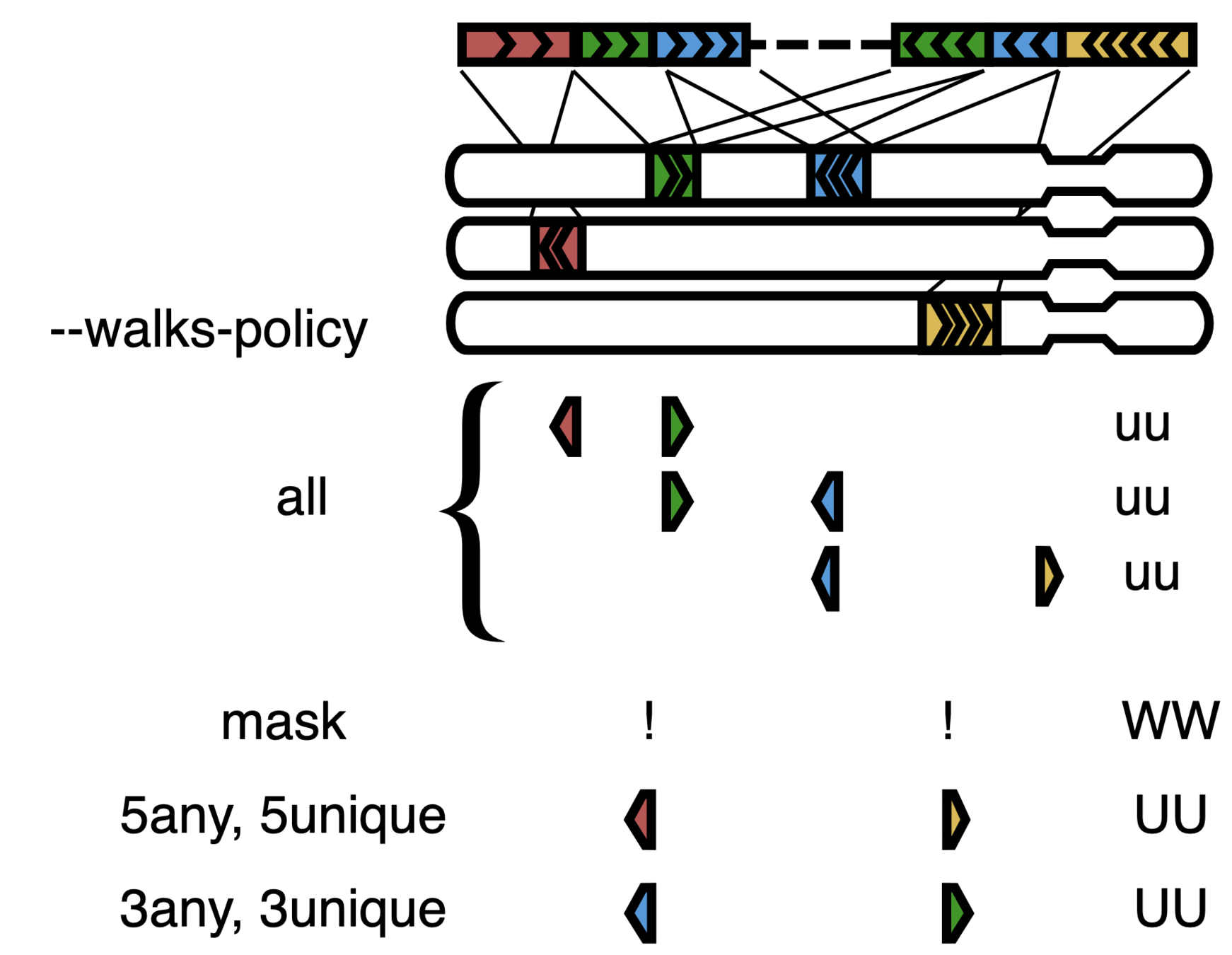
Алгоритм bwa mem позволяет картировать химеры:
Получение карты контактов
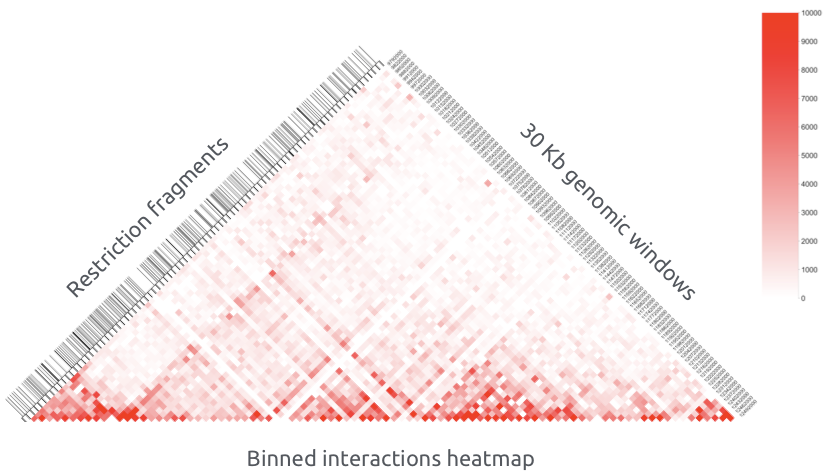
Бины генома - это последовательные окна геномных координат одинакового размера.
Каждая ячейка (или пиксель) контактной карты содержит количество всех контактов, пришедших рестриктных фрагментов, соответствующих таким бинам:
Бинированая карта контактов
Нормировка и корректировка Hi-C
Imakaev et al. Nature Methods 2012
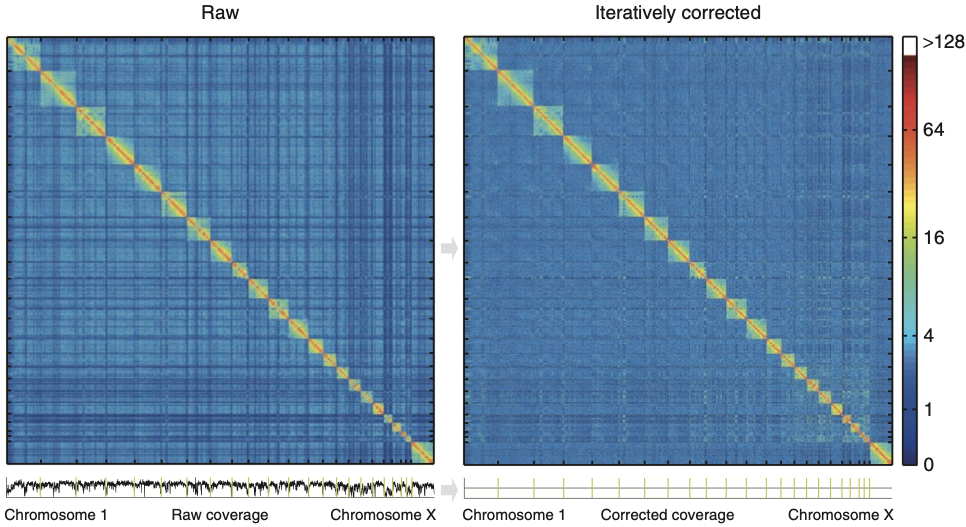
Нормировка матрицы Hi-C приводит матрицу к бистохастичному виду. Иными словами, выравнивается кумулятивная частота контактов регионов генома (Cumulative Contact Frequency, CCF):
Кумулятивная частота контактов участков генома
Samborskaya et al. PeerJ 2020
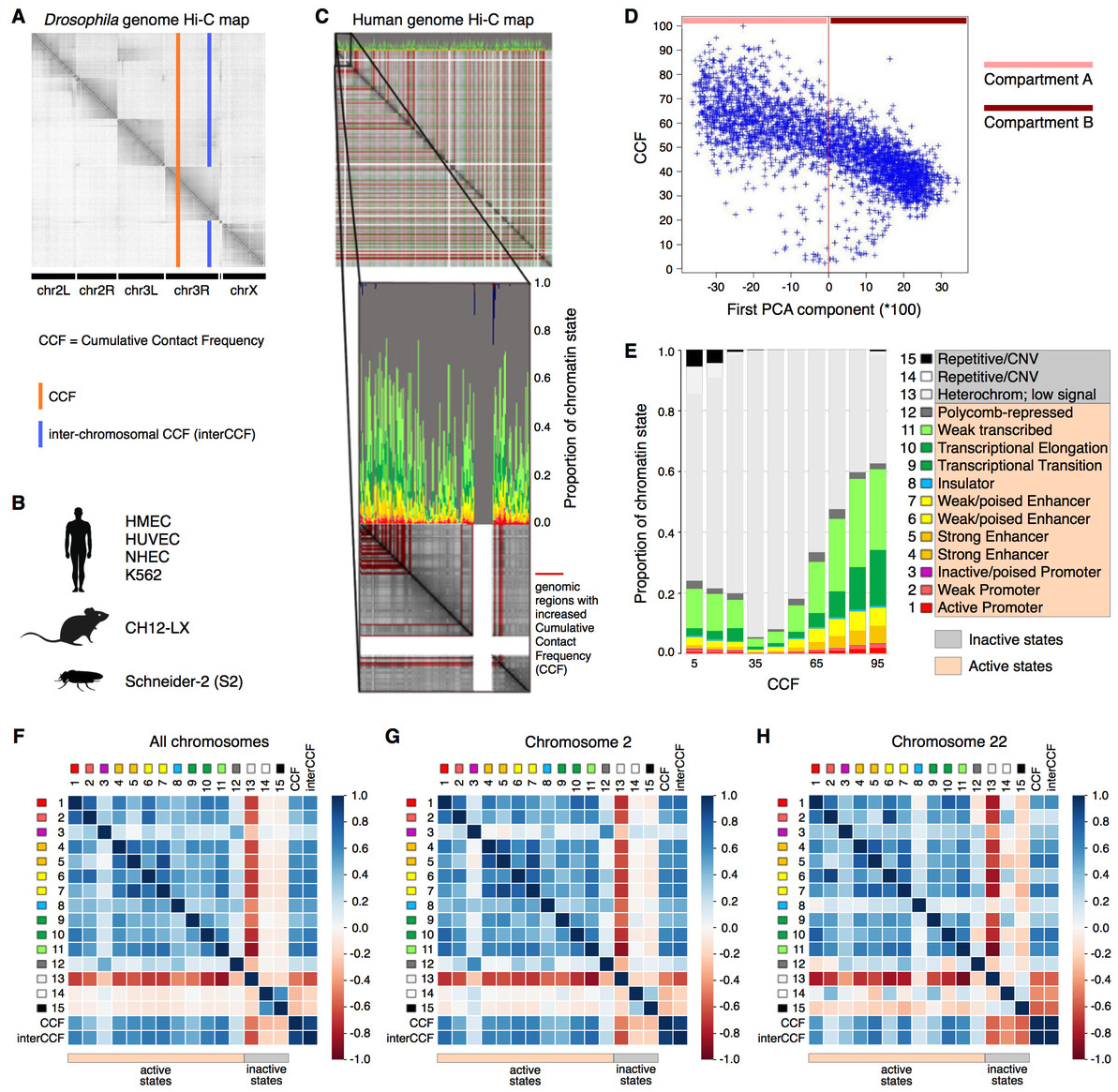
Подходы к корректировке Hi-C
Два основных типа:
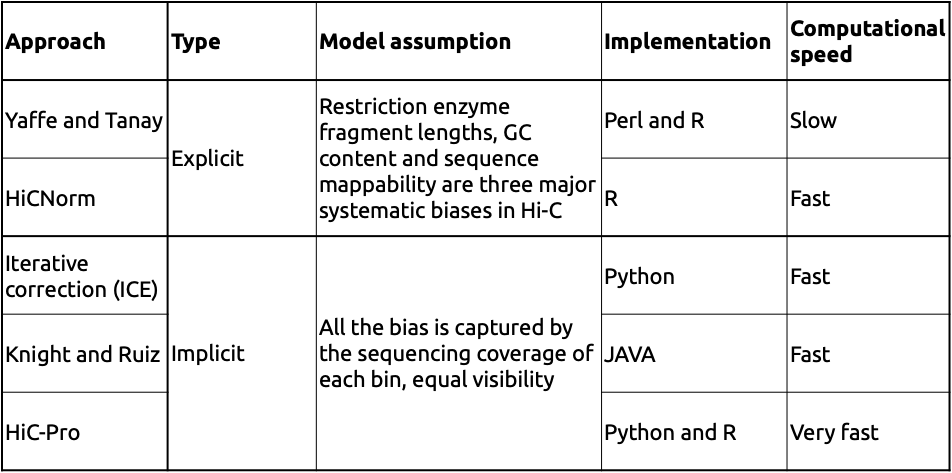
Adopted from Schmitt et al. Nature Reviews 2016
Примеры корректировки карты Hi-C

Schmitt et al. Nature Reviews 2016
Графики P(s): важный контроль качества данных
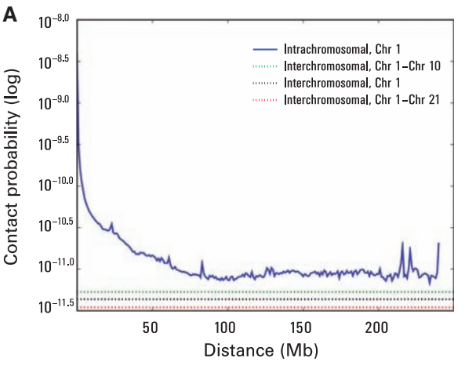
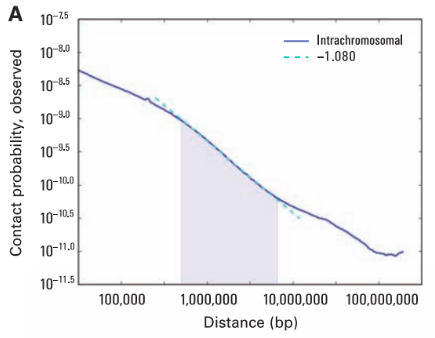
Lieberman-Aiden, 2009
Вероятность контакта хроматина
между двумя участками
зависит от геномного расстояния между ними
P(s)-график
log-log P(s)-график
P(s) и фрактальная размерность
Sanborn et al. PNAS 2015
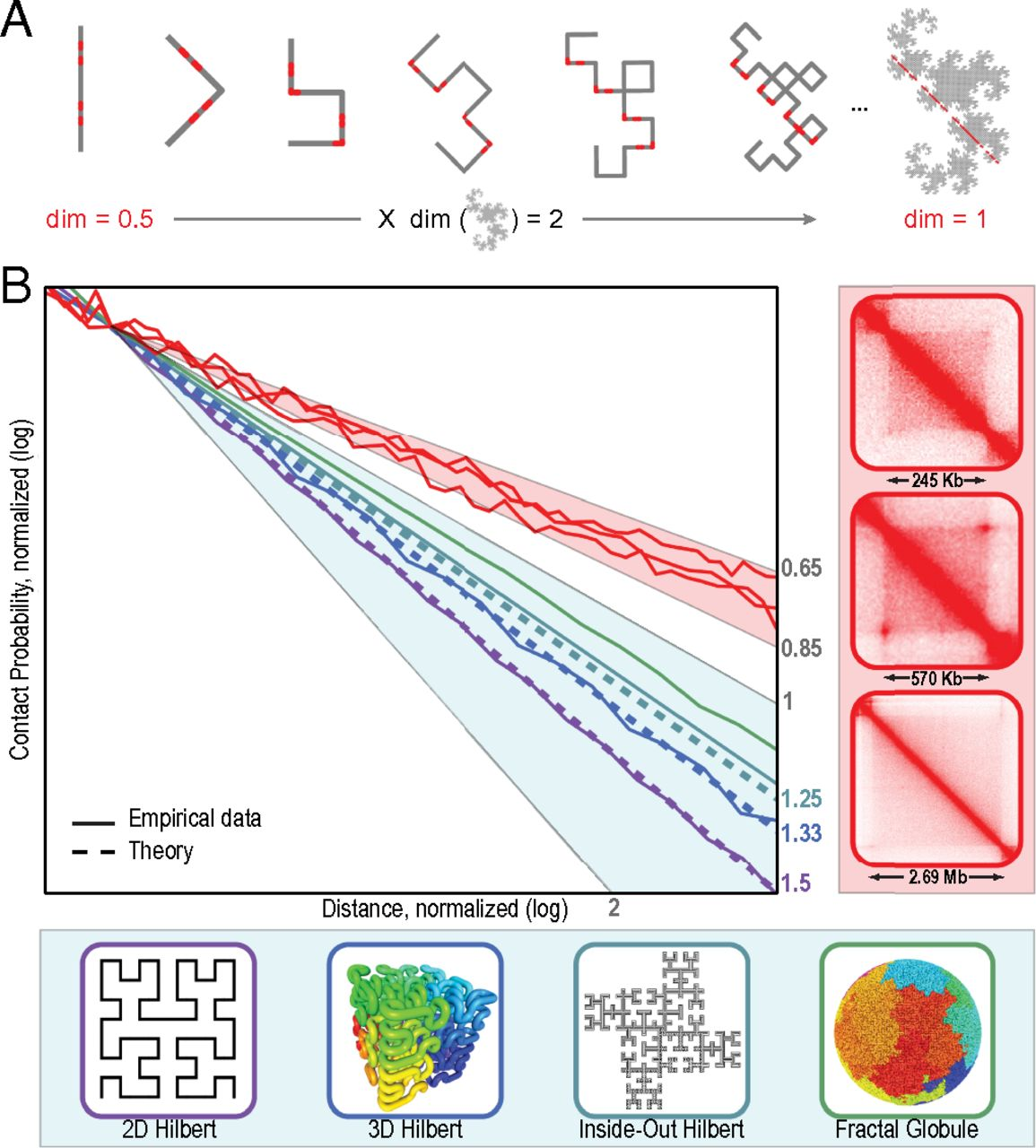
P(s) и фрактальная размерность полимера ДНК
Lieberman-Aiden et al. 2009
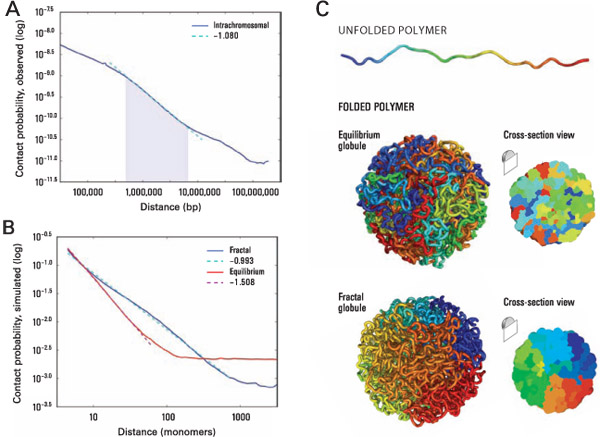
Фрактальная глобула - модель, близко описывающая наблюдаемые свойства P(s):
Когда Hi-C карты не подчиняются закону P(s)?
Oddes 2018
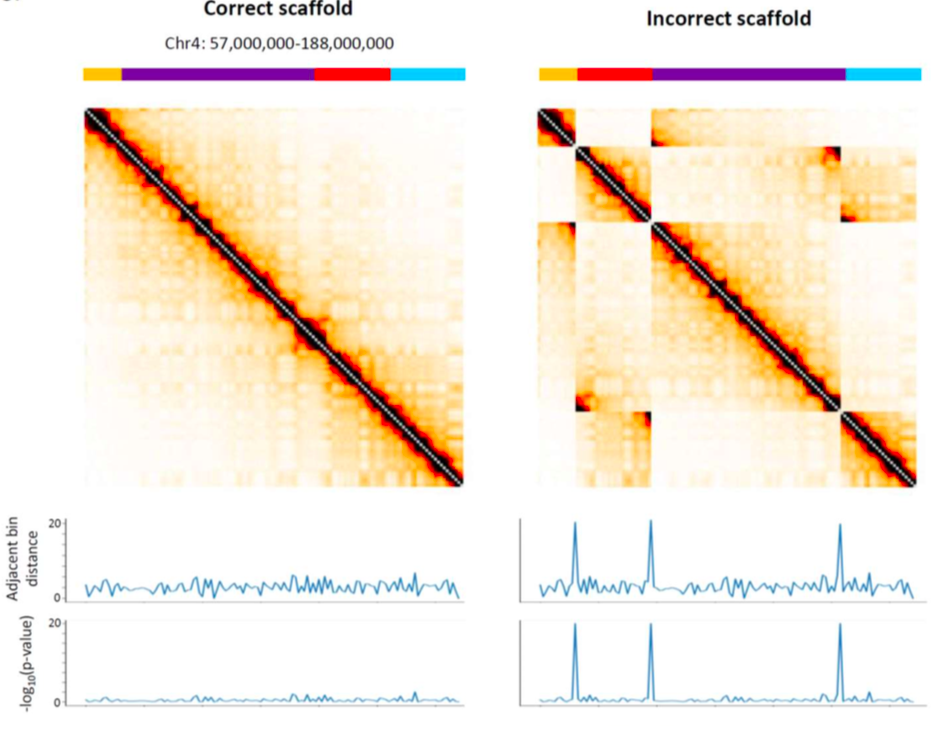
Закон P(s) нарушен?
Или что-то пошло не так с Hi-C?
Правильная последовательность ДНК (сборка)
Ошибка сборки: ДНК устроена не так, как мы думали!
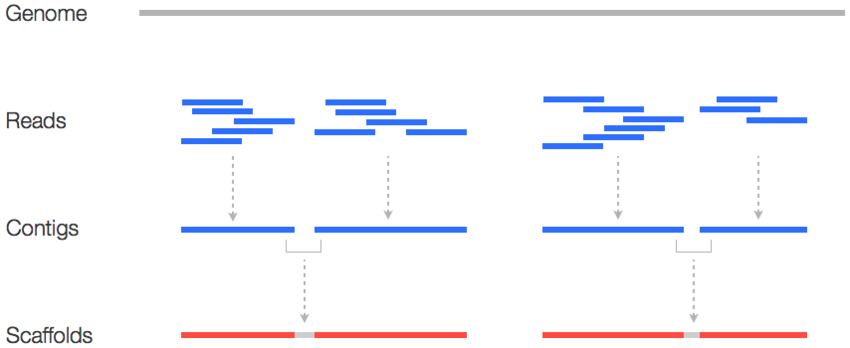
Решение задачи правильной сборки
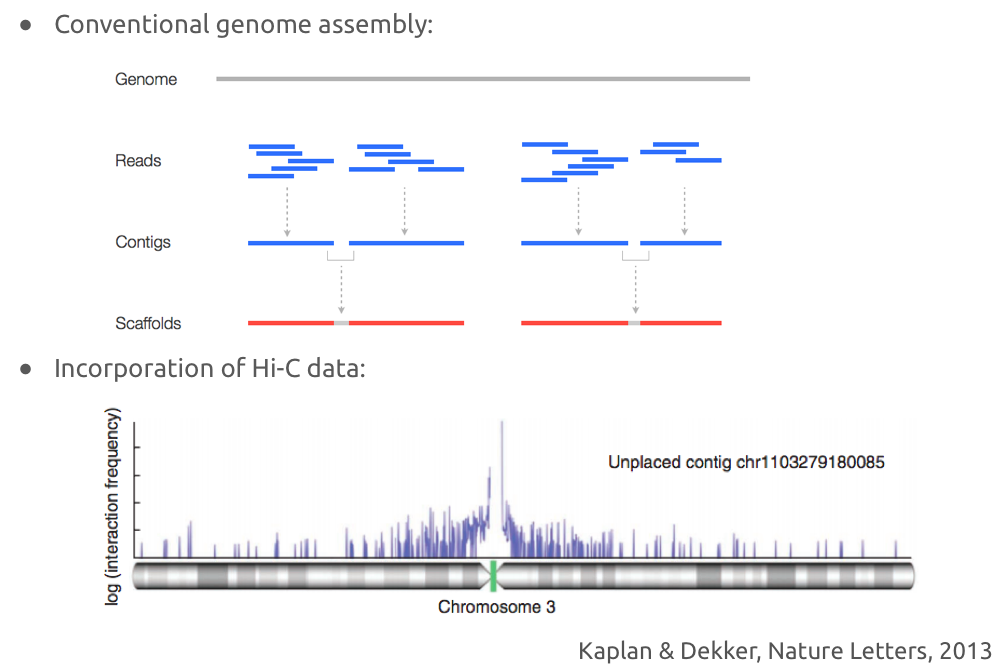
Обычная сборка генома по ридам ДНК:
Улучшение сборки с помощью Hi-C:
Пример успешного решения задачи сборки с помощью Hi-C:
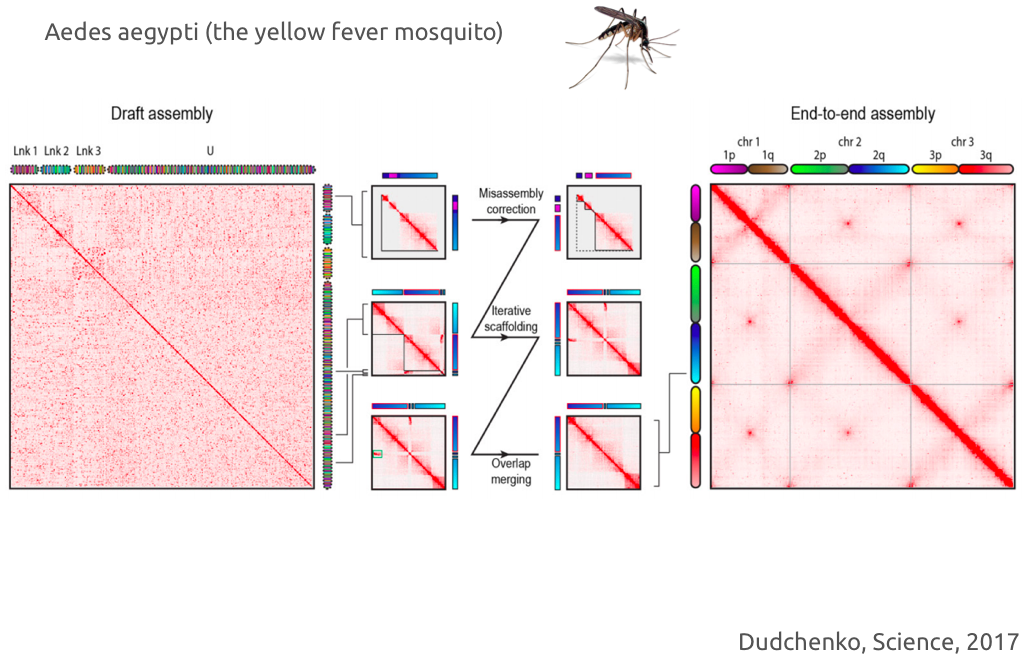
Пример использования: https://youtu.be/IMmVp8FodmY
Cборка геномов разнообразных организмов с помощью Hi-C
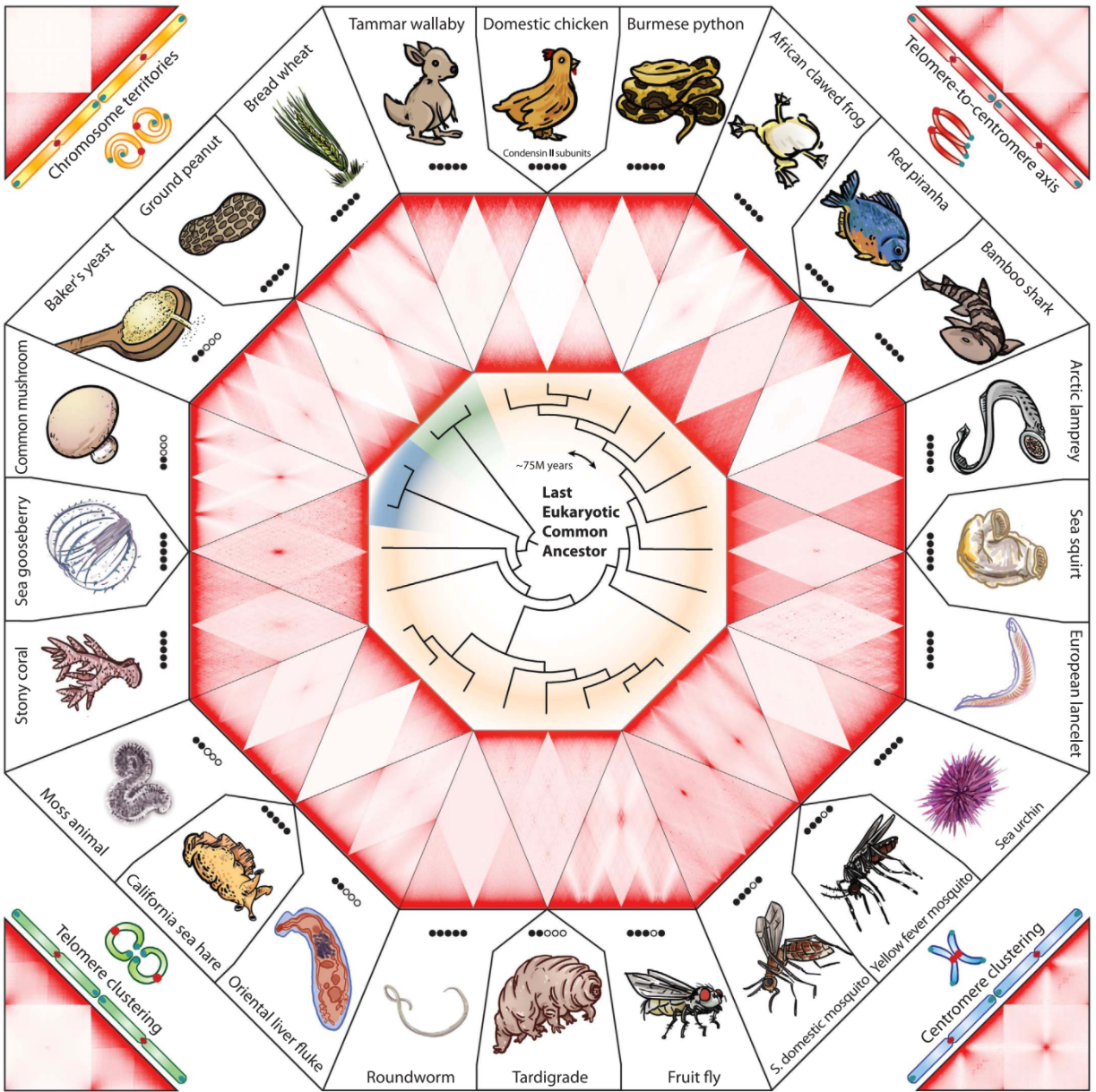
Структура хроматина в разных царствах организмов:
Обработка данных Hi-C
0. Контроль качества данных NGS
- Программа: fastqc
- Выдача: html-репорт
1. Картирование данных парноконцевого NGS
- Программа: bwa
- Выдача: sam-файл
2. Получение пар контактов Hi-C и их дедупликация
- Программа: pairtools
- Выдача: pairs-файл
3. Бинирование и нормализация
- Программа: cooler
- Выдача: cool-файл
4. Контроль качества
- Программа: multiqc
- Выдача: html-репорт
Руководство к анализу Hi-C
Картирование данных
- Картирование, переводит
fastq (файл с ридами) в sam (файл с выравниваниями):
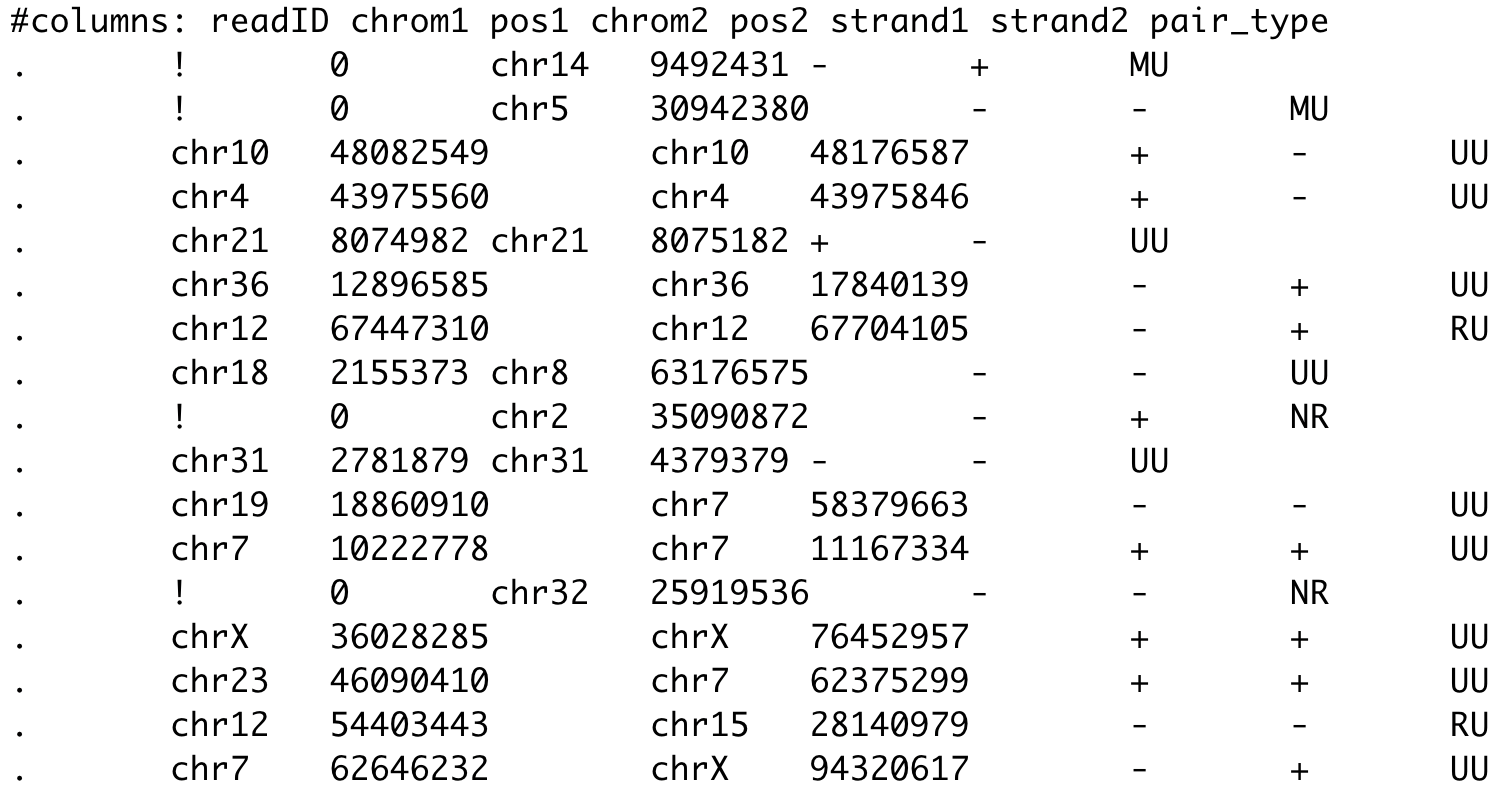
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam}$ pairtools parse -c ${chromosome_sizes_file} ${input.sam} -o ${output.pairs} \
--drop-seq --drop-sam --drop-readid --min-mapq 30- Чтение sam в pairs (файл с парами контактов pairs):
Картирование данных
- Картирование, переводит
fastq (файл с ридами) в sam (файл с выравниваниями):
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam}$ pairtools parse -c ${chromosome_sizes_file} ${input.sam} -o ${output.pairs} \
--drop-seq --drop-sam --drop-readid --min-mapq 30- Чтение sam в pairs (файл с парами контактов pairs):
$ pairtools dedup --output-stats ${output.dedup.stats} ${output.pairs} \
-o ${output.nodups.pairs}- Дедупликация pairs-файла:
Контроль качества пар
- Запуск multiqc на выдаче описательной статистики:
multiqc --module pairtools ./- Файл с описательной статистикой был на предыдущем шаге:
$ pairtools stats -o ${output.stats} ${output.nodups.pairs}Контроль качества Hi-C

Типы картирований:
Контроль качества пар
Разные типы пар контактов:
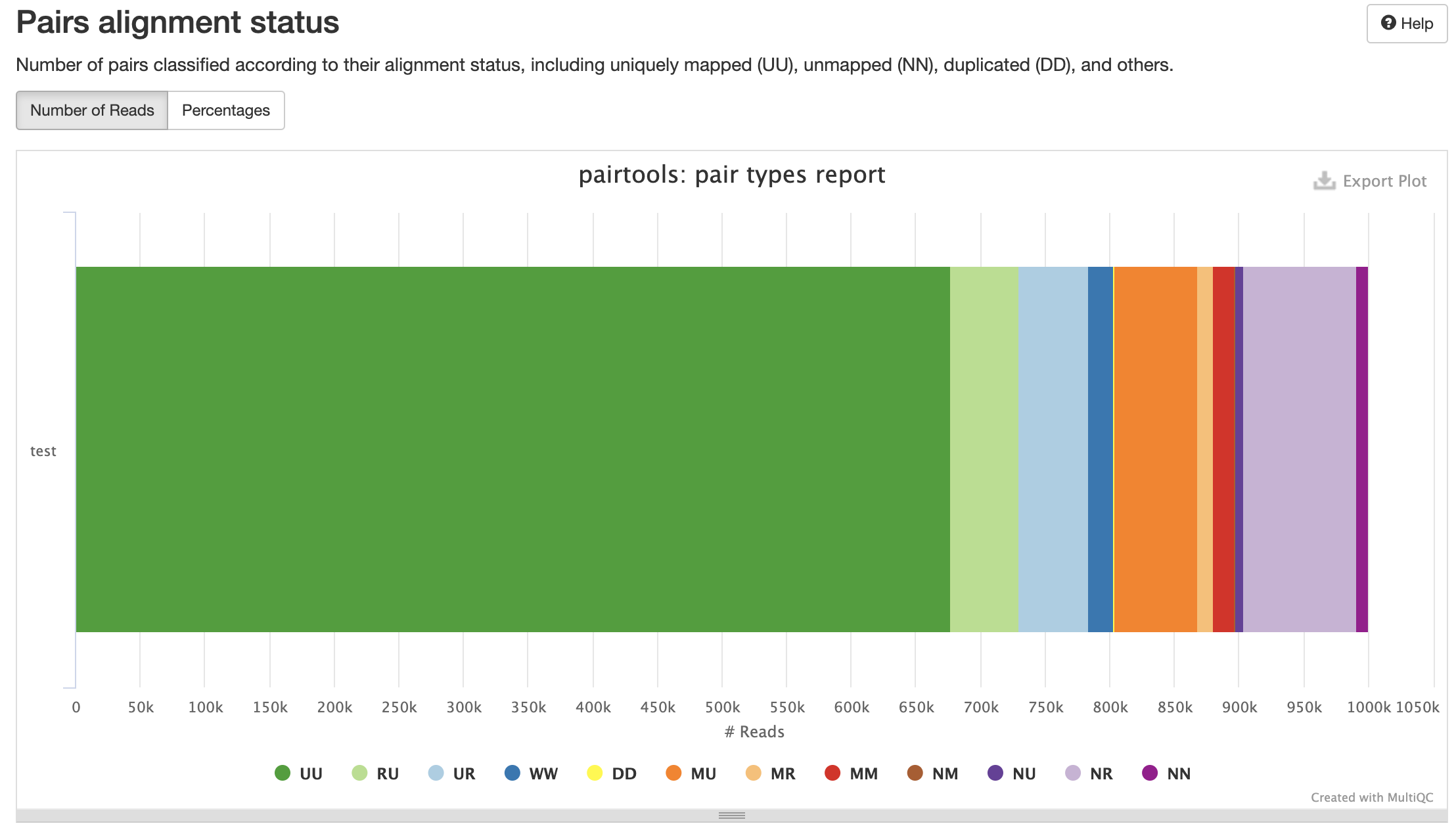
Контроль качества пар
Количество контактов для разного геномного расстояния:
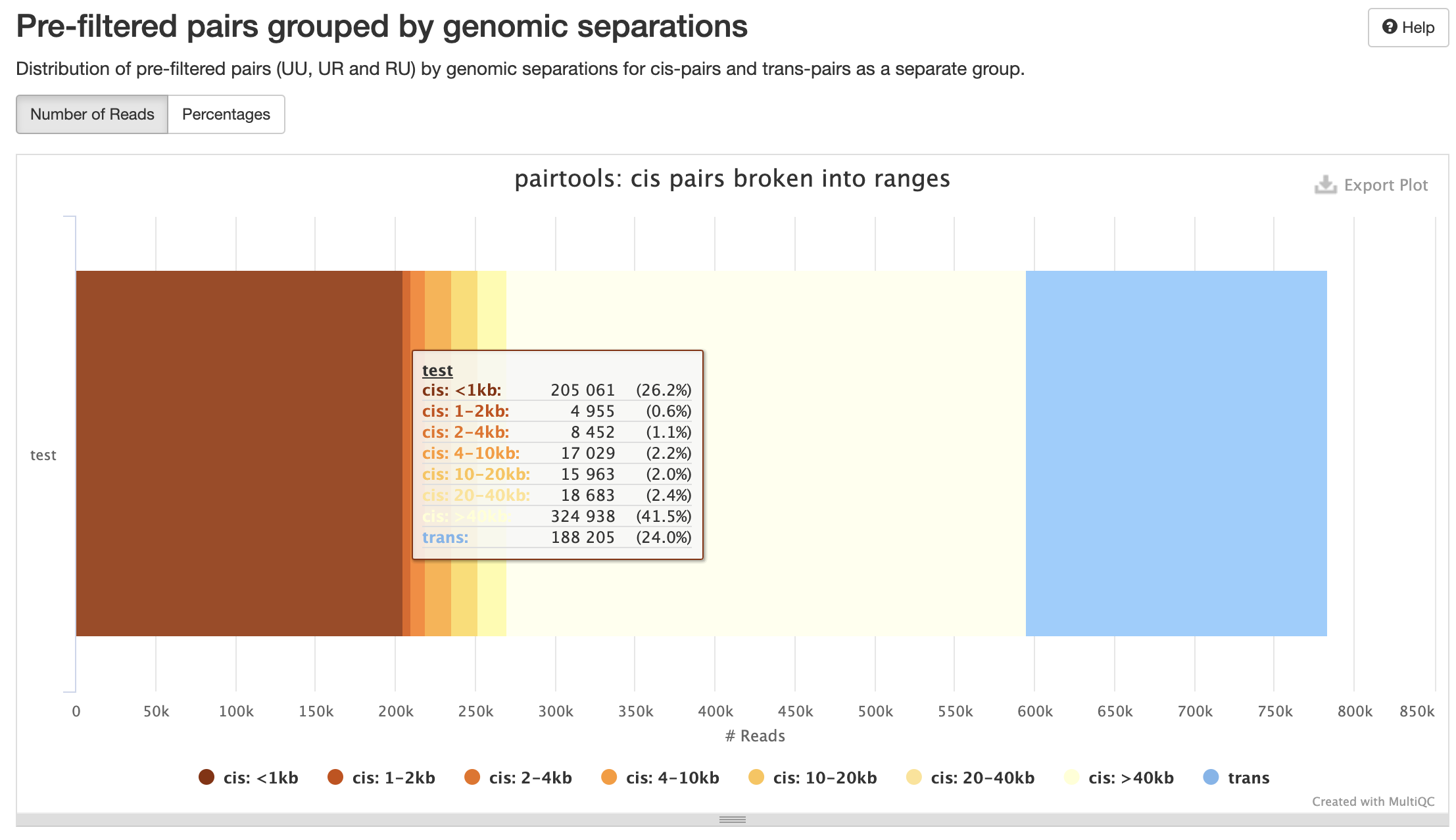
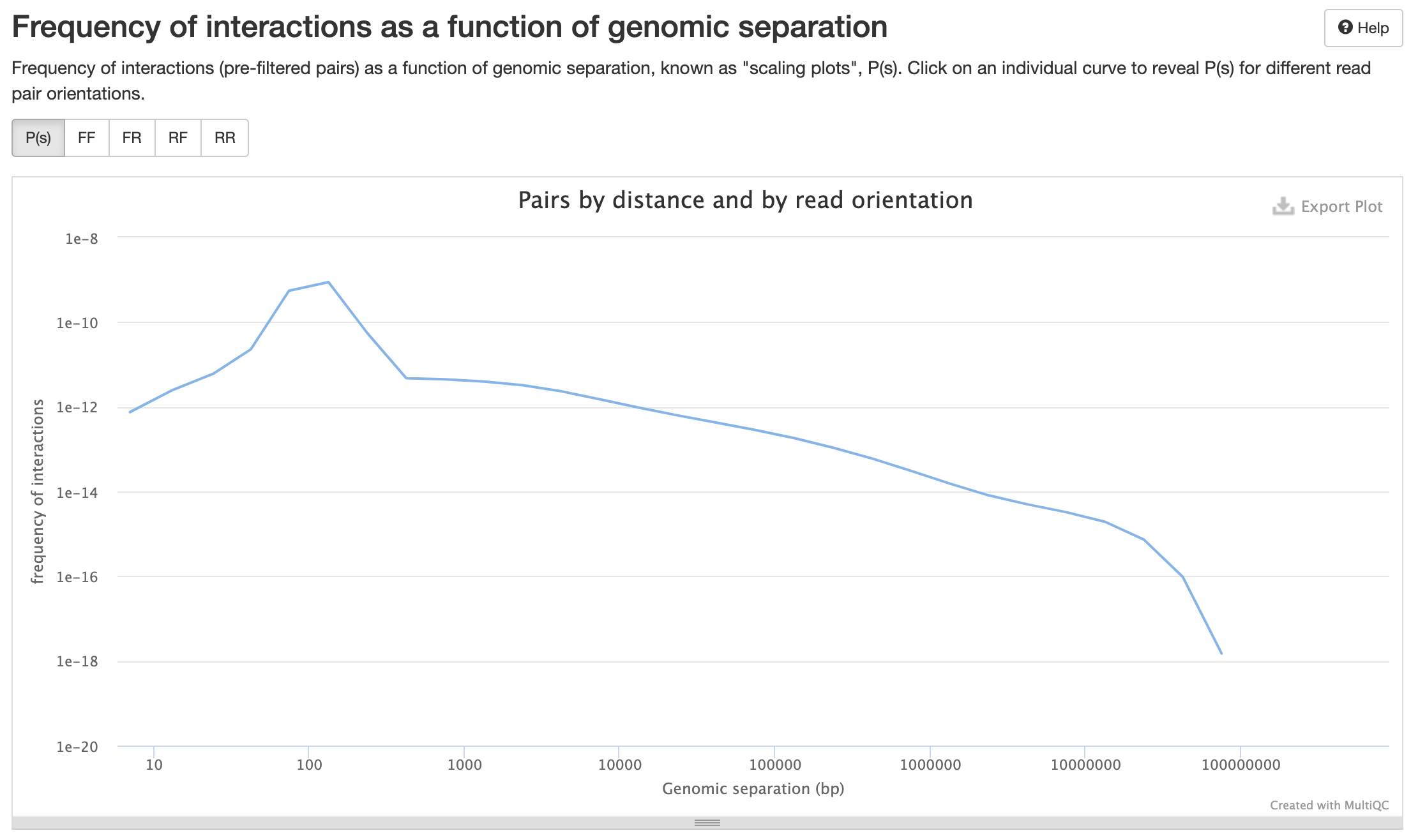
Контроль качества пар
P(s):
Получение контактных карт
$ cooler cload pairs -c1 2 -c2 4 -p1 3 -p2 5 ${chromsizes}:1000000 ${input.nodup.pairs} ${out.cool}- Загрузка контактных пар в cool-файл:
$ cooler balance ${out.cool}- Проводим корректирование Hi-C данных:
$ cooler info ${out.cool}- Проверяем запись и информацию:
(*) 100000 - это размер одного бина Hi-C карты, иначе говоря, 100 Kb
Попробуйте разные разрешения контактных карт для визуализации. На бактериях можно использовать размер бина 5-10 Kb, на крупных геномах - от 50 Kb до 1 Mb.
Визуализация карт
$ cooler show ${in.cool} chr1 -o ${image.png}- Просмотр одной хромосомы:
- Для просмотра полного генома воспользуйтесь HiCExplorer:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicPlotMatrix.html#hicplotmatrix
Попробуйте разные параметры визуализации:
тип шкалы (log or linear),
максимальное и минимальное значение и прочие.
Можно выбрать разное разрешение карт на предыдущем шаге.
Задача: добиться лучшей визуализации структурных элементов хроматина
Заключительные ремарки
Задание
Установка: вы работаете в биоинформатической лаборатории по исследованию эволюционных процессов в хроматине. Вам даны:
- данные Hi-C для некоторого организма,
-
база данных о геномах с аннотацией видов
Ваши задачи:
1) определить организм, для которого выполнен Hi-C;
2) провести процедуру обработки данных Hi-C;
3) вынести вердикт: хорошо ли сработал протокол Hi-C и секвенирование
4) провести интерпретацию результата и предложить гипотезы об особенностях укладки хроматина организма
Форма для ответов:
https://forms.gle/dqBa4ENySyCywD2T6
Максимально 10 баллов за ответ. Ошибки можно компенсировать за счет последнего задания на биологическую интерпретацию результатов.
Дедлайн: 9 декабря в 11:10
(будем разбирать правильные ответы!)
Кластер
1. Логин через терминал:
или Putty для Windows.
(инструкции: https://www.ssh.com/ssh/putty/windows/#sec-Configuration-options-and-saved-profiles)
2. Активируйте удобный bash-терминал:
3. Создайте директорию проекта:
4. Настройка рабочей среды и ее проверка:
$ ssh -p5222 username@92.242.58.92 или ssh -p32222 username@92.242.58.92$ mkdir practice_chromatin
$ cd practice_chromatin$ /usr/share/data-minor-bioinf/chromatin/anaconda/condabin/conda init
$ conda activate chromatin
$ cooler --version$ bashДанные
- База данных геномов:
- Аннотация базы данных геномов:
https://docs.google.com/spreadsheets/d/1lhHjgq2HT83RCr_EBU9IyD2iQ7paUIHG-5Qh95pLd28/edit?usp=sharing
3. База данных секвенирования:
4. Ваш уникальный идентификатор данных:
https://docs.google.com/spreadsheets/d/1rJA3Cz1itIv8Z_cBjMaBK1vpKnYe7kOKzGrsrCmrw9E/edit?usp=sharing
ls /mnt/storage/chromatin/genomes/В папках содержится информация:
1. Прочтения секвенирования, первые и вторые из пар:
/mnt/storage/chromatin/fastq/<YOUR CODE>_R1.fastq
/mnt/storage/chromatin/fastq/<YOUR CODE>_R2. fastq
2. Файл с размерами хромосом данного организма:
/mnt/storage/chromatin/genomes/<YOUR GENOME ID>/<YOUR GENOME ID>.fa.sizes
3. Индекированный геном организма:
/mnt/storage/chromatin/genomes/<YOUR GENOME ID>/index/bwa/<YOUR GENOME ID>.fa*
ls /mnt/storage/chromatin/fastq/