A briefer history of
bcbio-nextgen(-vm)
I. Molecular biology
II. Human genome
III. bcbio-nextgen
IV. bcbio-nextgen-vm
Table of content
Disclaimer

I am NOT a
scientist
I. Amino Acids
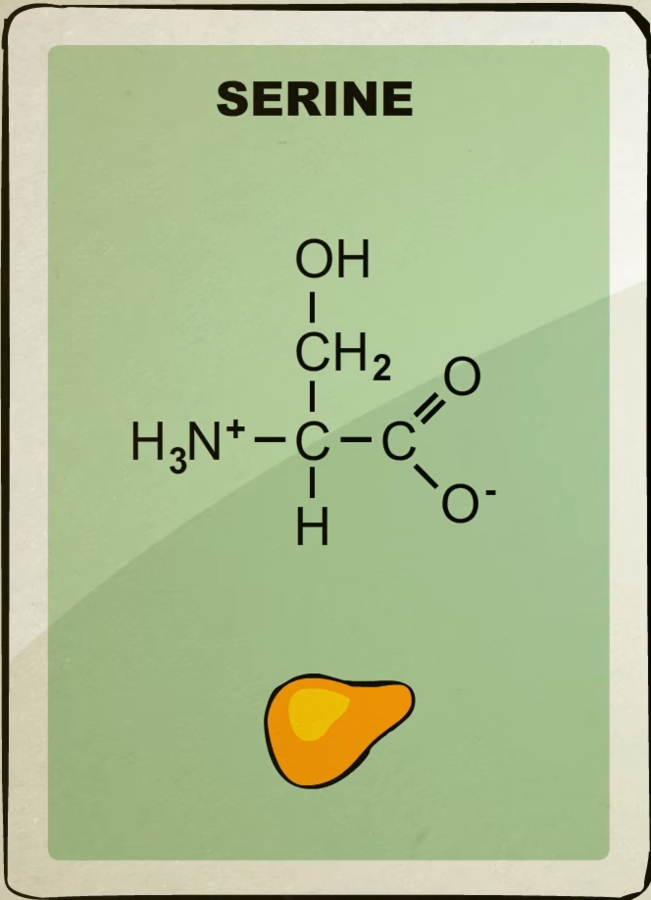
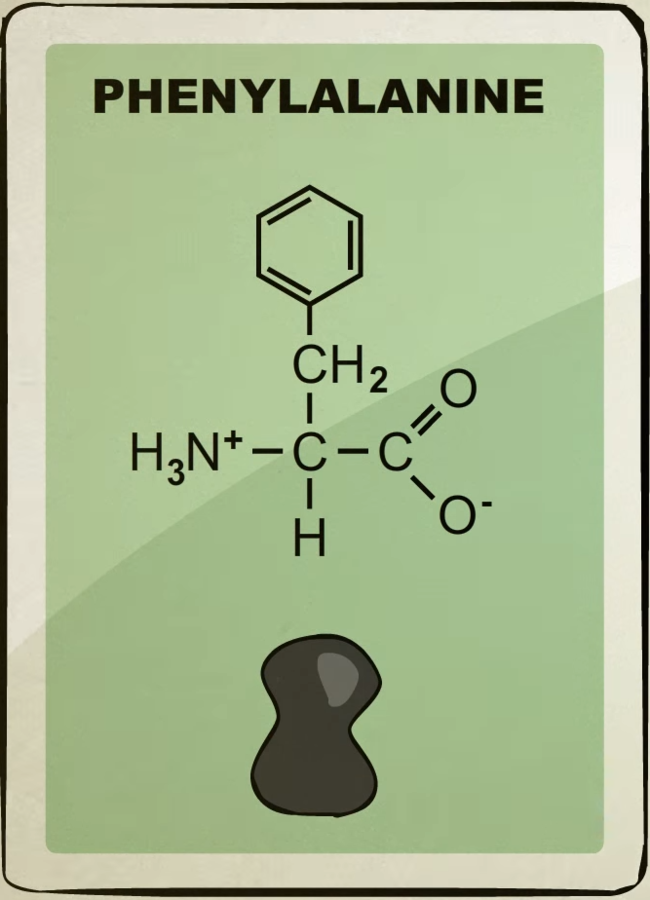
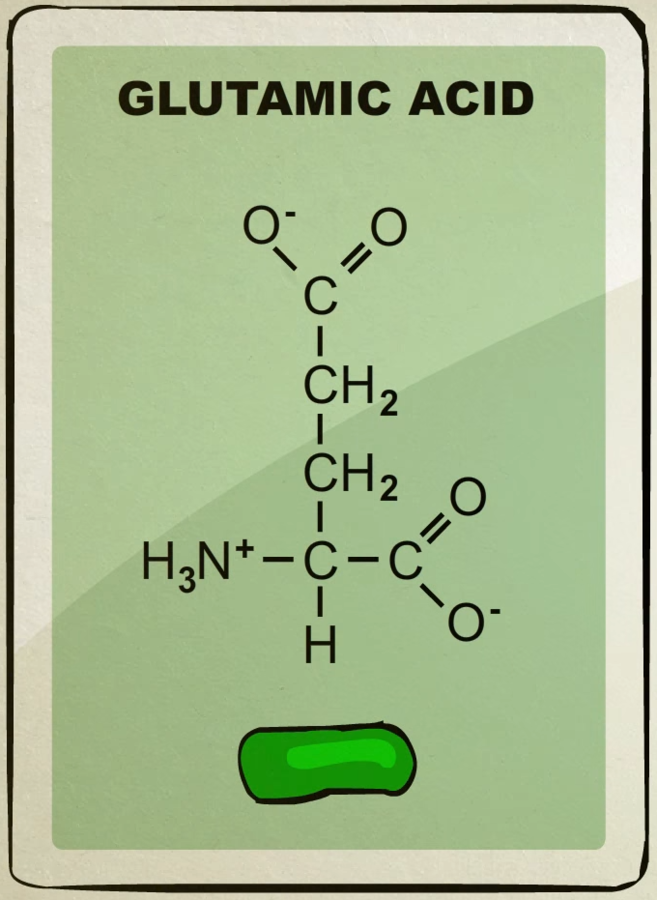
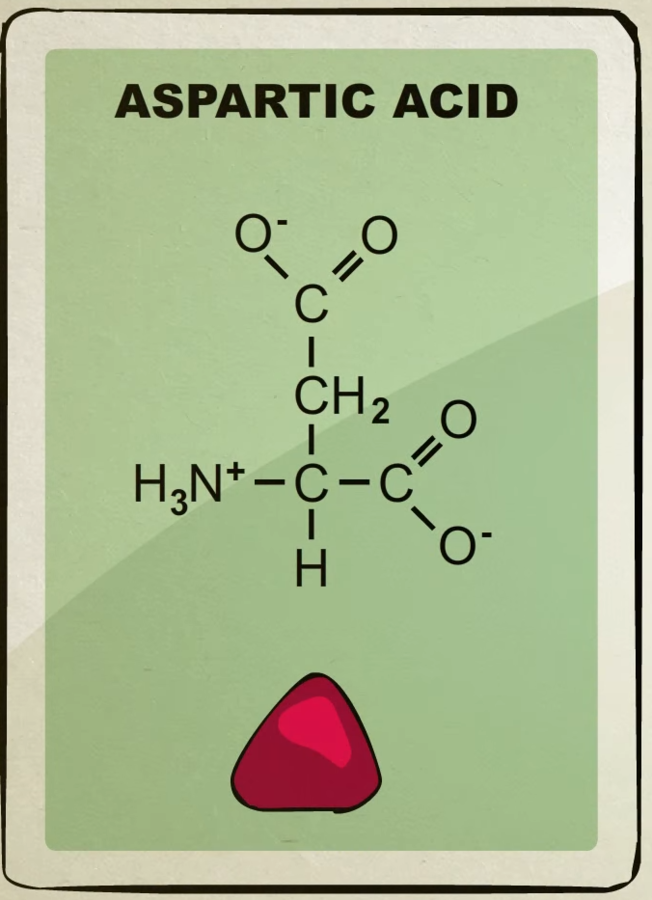
There’s about 20 different kinds of amino acids each with their own unique shape.
II. Human genome

GRCh38.p3 (Genome Reference Consortium Human, Build 38)
www.ensembl.org/Homo_sapiens/Location/Genome
II. Mutation

I am P53 the protector of cells !
Who am I ?
II. Time to solve some diseases



Configuration
Variations
Insertions
Deletions
SNPs
Quality
Alignment
Coverage
Analisys
Annotations
Query
III. bcbio-nextgen
III. Utilities
bedtools, bcftools, biobambam, sambamba, samblaster, samtools, vcflib
Aligners:
bwa-men, novoalign, bowtie2
Variantion:
FreeBayes, GATK, Platypus, MuTecT, scalpel, SnpEff, VEP, GEMINI, Lumpy, Delly
RNA-seq:
Tophat, STAR, cufflinks, HTSeq
Quality control:
fastqc, bamtools, RNA-SeQC
Manipulation:
III. Users
- Harvard School of Public Health
- Massachusetts General Hospital
- Science for Life Laboratory
- Institute of Human Genetics, UCSF
- IRCCS “Mario Negri” Institute for Pharmacological Research
- The Translational Genomics Research Institute (TGen)
- Computer Science and Artificial Intelligence Laboratory (CSAIL), MIT
More information can be found on the following link:
https://bcbio-nextgen.readthedocs.org/en/latest/contents/introduction.html
III. How to run it ?


bcbio-nextgen-vm

IV. Elasticluster
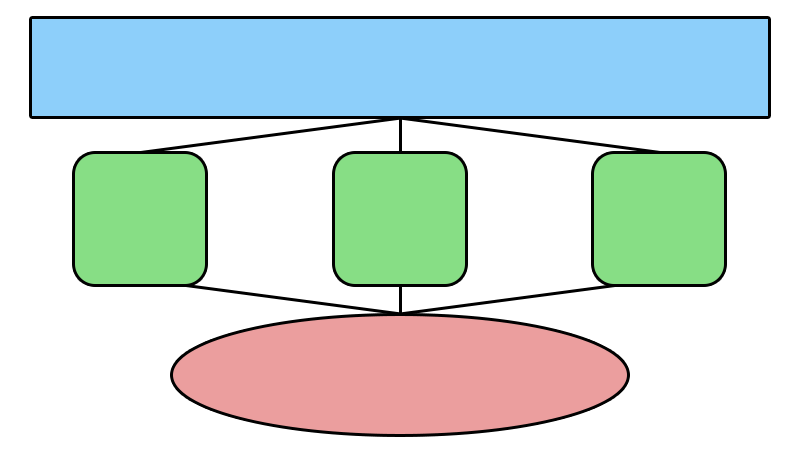
Front Node
Compute
Node
Compute
Node
Compute
Node
Messaging
IV.
Ansible is a radically simple IT automation platform that makes your applications and systems easier to deploy.

IV. Docker
Docker containers wrap up a piece of software in a complete filesystem that contains everything it needs to run: code, runtime, system tools, system libraries - anything you can install on a server.
IV. SLURM

"My general thoughts are that this is an exciting time to contribute to open source work in bioinformatics.
This will enable individuals to learning more about their genetics and tailor their lifestyles and disease management. In terms of open source bioinformatics research, this means we'll have an increasing pool of individuals interested in being able to analyze their own genomes.
We hope that this will enable everyone to investigate their own genomes and build a large community of both professional and amateur scientists."

Brad Chapman
Research Associate, Bioinformatics Core la Harvard School of Public Health

Questions !?
Resources
- Documentation: readthedoc.org/bcbio-nextgen
- GitHub:
- Ansible: Documentation, Resources
- Stated Clearly: What is a gene?
- Stated Clearly: What is DNA and How Does it Work?