Constant pH
What am I possibly hoping to achieve?
Bas Rustenburg // Chodera lab
#pHallthetime
Stardate 93387.66
Disclaimer
For the purpose of this presentation, I've experimented with multiple ways to provide a more interactive experience. Feedback is welcome.
Live web-cast
Follow the slides live on your own computer using this url:
#pHallthetime
http://slides.com/basrustenburg/deck/live
Why am I interested in constant-pH work?
Protonation state effects
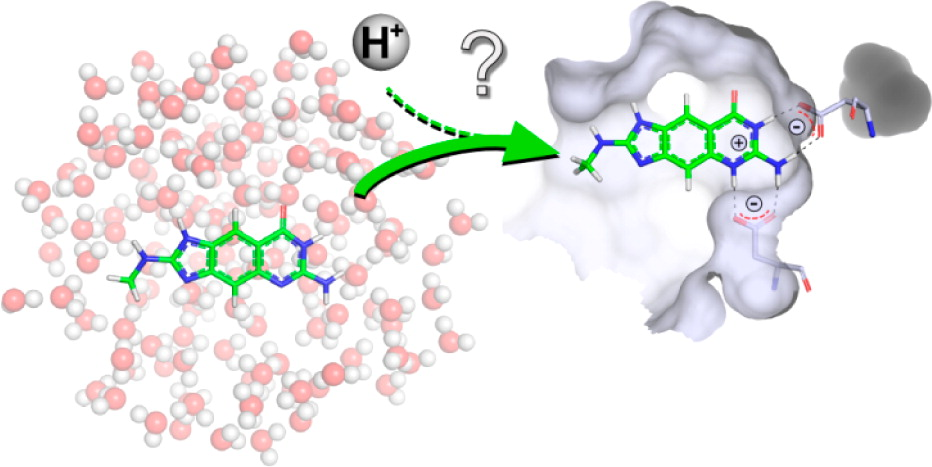
Neeb et al. J. Med. Chem., 2014, 57 (13), pp 5554–5565
There is no a priori way to know every relevant protonation state of a ligand or protein, and how relevant they are to the binding affinity.
The standard state approach
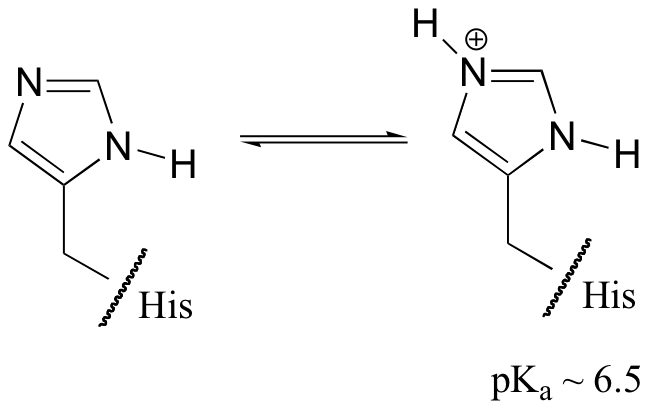
- Typically, one only simulates a single protonation state.
- For proteins at neutral pH, only histidine is closely looked at.
Problems that...
- If reality has a mixture of protonation states, we're ignoring their contributions to the free energy of the system
- If the most relevant protonation state changes upon binding, we simulate the wrong state
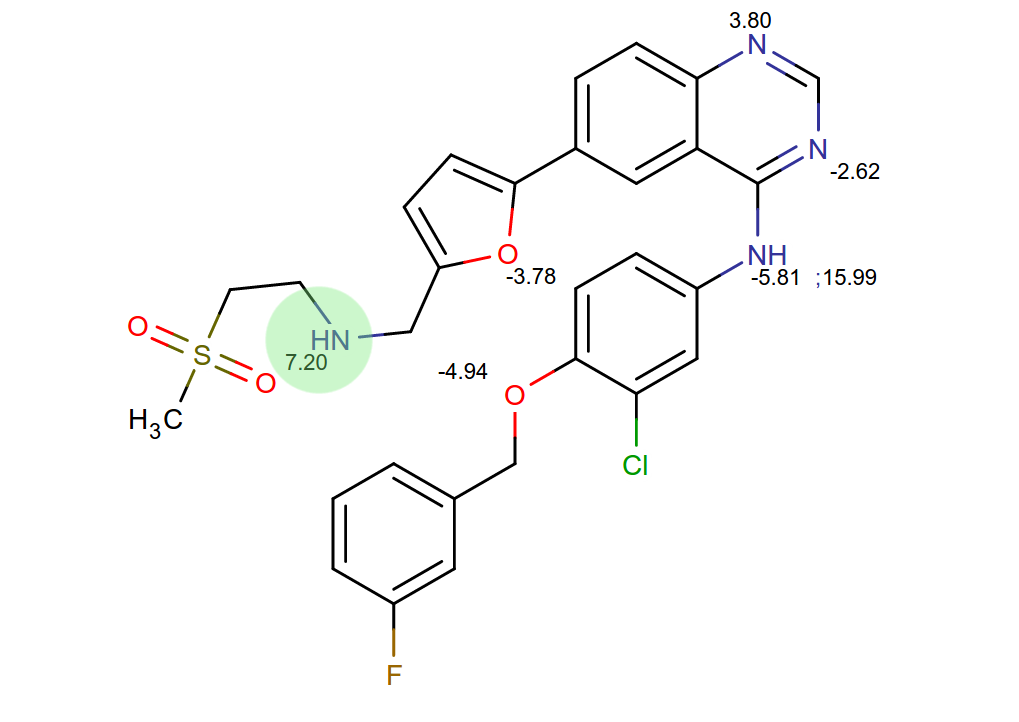
Bonus question
- What kinase inhibitor is this?
It's....
Lapatinib!
#ofcourseitslapatinib
The standard approach is ignoring contributions of unknown magnitude, for imatinib and Abl there is clear indication of the relevance
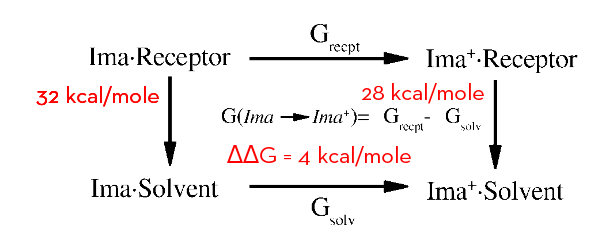
Aleksandrov, A and Simonson, T J Comput Chem 31,7, pp. 1550–1560 (2010)
More problems with the Standard aproach
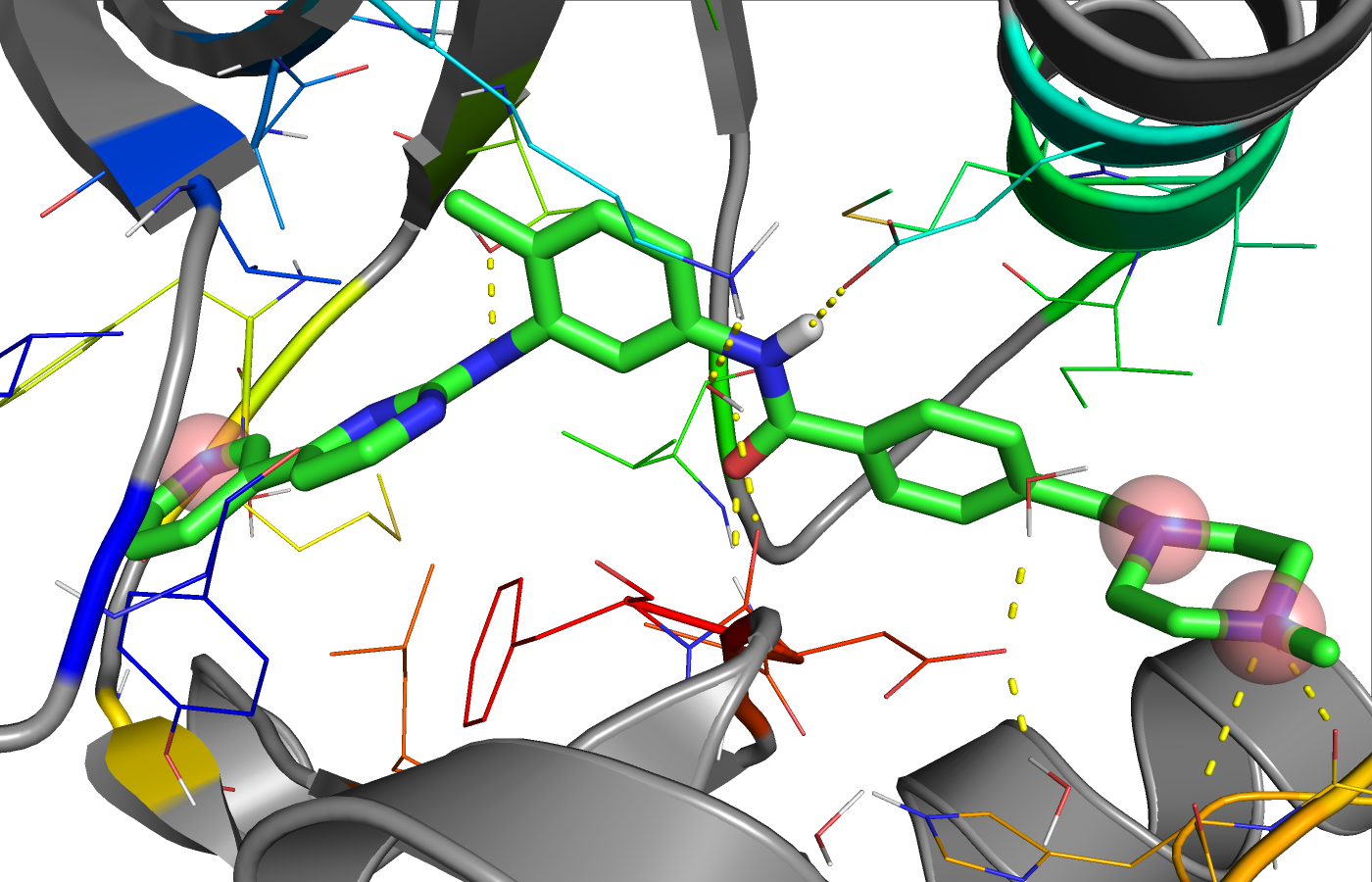
Imatinib bound to Abl kinase
Kinase inhibitors
pKa predictions using EPIK suggest that many kinase inhibitors have more than one accessible protonation state at pH 7.4

Figure by: John & Julie
What is...
constant-pH?
#pHallthetime
And how does it solve our problems?
Constant PH
Allow for the exchange of protons with an external proton "bath", which maintains a constant pH.
Constant PH
In implicit solvent
- Take a reference state with a known pKa, such as a free dipeptide or capped peptide in solution
- ΔGelec difference between the electrostatic potential for the current protonation state and proposed state
Mongan, Case, and McCammon • Journal of Computational Chemistry
Volume 25, Issue 16, pages 2038–2048, December 2004
Constant PH
In implicit solvent
- (ΔGelec) take difference between the electrostatic potential for the current protonation state and proposed state
- Accept using
Mongan, Case, and McCammon • Journal of Computational Chemistry
Volume 25, Issue 16, pages 2038–2048, December 2004
Openmm-ConstpH
- Currently works for implicit solvent only
- Systems are prepared using Ambertools, including tleap, cpinutils
- Residue names need to be predefined (tough for ligands)
- Have you ever seen a cpinfile?
https://github.com/choderalab/openmm-constph
#pHallthetime
Openmm-ConstpH
Example of a cpin file
&CNSTPH
CHRGDAT=-0.4157,0.2719,0.0145,0.0779,-0.0398,-0.0173,-0.0173,0.0136,-0.0425,
-0.0425,0.8054,-0.8188,-0.8188,0.0,0.5973,-0.5679,0.0,0.0,0.0,-0.4157,0.2719,
0.0145,0.0779,-0.0071,0.0256,0.0256,-0.0174,0.043,0.043,0.6801,-0.5838,-0.6511,
0.4641,0.5973,-0.5679,0.0,0.0,0.0,-0.4157,0.2719,0.0145,0.0779,-0.0071,0.0256,
0.0256,-0.0174,0.043,0.043,0.6801,-0.5838,-0.6511,0.0,0.5973,-0.5679,0.4641,
0.0,0.0,-0.4157,0.2719,0.0145,0.0779,-0.0071,0.0256,0.0256,-0.0174,0.043,0.043,
0.6801,-0.6511,-0.5838,0.0,0.5973,-0.5679,0.0,0.4641,0.0,-0.4157,0.2719,0.0145,
0.0779,-0.0071,0.0256,0.0256,-0.0174,0.043,0.043,0.6801,-0.6511,-0.5838,0.0,
0.5973,-0.5679,0.0,0.0,0.4641,-0.3479,0.2747,-0.24,0.1426,-0.0094,0.0362,
0.0362,0.0187,0.0103,0.0103,-0.0479,0.0621,0.0621,-0.0143,0.1135,0.1135,
-0.3854,0.34,0.34,0.34,0.7341,-0.5894,-0.3479,0.2747,-0.24,0.1426,-0.10961,
0.034,0.034,0.06612,0.01041,0.01041,-0.03768,0.01155,0.01155,0.32604,-0.03358,
-0.03358,-1.03581,0.0,0.38604,0.38604,0.7341,-0.5894,-0.4157,0.2719,0.0341,
0.0864,-0.1783,-0.0122,-0.0122,0.7994,-0.8014,-0.8014,0.0,0.5973,-0.5679,0.0,
0.0,0.0,-0.4157,0.2719,0.0341,0.0864,-0.0316,0.0488,0.0488,0.6462,-0.5554,
-0.6376,0.4747,0.5973,-0.5679,0.0,0.0,0.0,-0.4157,0.2719,0.0341,0.0864,-0.0316,
0.0488,0.0488,0.6462,-0.5554,-0.6376,0.0,0.5973,-0.5679,0.4747,0.0,0.0,-0.4157,
0.2719,0.0341,0.0864,-0.0316,0.0488,0.0488,0.6462,-0.6376,-0.5554,0.0,0.5973,
-0.5679,0.0,0.4747,0.0,-0.4157,0.2719,0.0341,0.0864,-0.0316,0.0488,0.0488,
0.6462,-0.6376,-0.5554,0.0,0.5973,-0.5679,0.0,0.0,0.4747,-0.4157,0.2719,
-0.0014,0.0876,-0.0152,0.0295,0.0295,-0.0011,-0.1906,0.1699,-0.2341,0.1656,
0.3226,-0.5579,0.3992,-0.2341,0.1656,-0.1906,0.1699,0.5973,-0.5679,-0.4157,
0.2719,-0.0014,0.0876,-0.0858,0.019,0.019,-0.213,-0.103,0.132,-0.498,0.132,
0.777,-0.814,0.0,-0.498,0.132,-0.103,0.132,0.5973,-0.5679,
PROTCNT=0,1,1,1,1,3,2,0,1,1,1,1,1,0,
RESNAME='System: Unknown','Residue: GL4 7','Residue: LYS 13','Residue: AS4 18',
'Residue: TYR 20','Residue: TYR 23','Residue: LYS 33','Residue: GL4 35',
'Residue: AS4 48','Residue: AS4 52','Residue: TYR 53','Residue: AS4 66',
'Residue: AS4 87','Residue: LYS 96','Residue: LYS 97','Residue: AS4 101',
'Residue: LYS 116','Residue: AS4 119',
RESSTATE=0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,
STATEINF(0)%FIRST_ATOM=102, STATEINF(0)%FIRST_CHARGE=0,
STATEINF(0)%FIRST_STATE=0, STATEINF(0)%NUM_ATOMS=19, STATEINF(0)%NUM_STATES=5,
STATEINF(1)%FIRST_ATOM=187, STATEINF(1)%FIRST_CHARGE=95,
STATEINF(1)%FIRST_STATE=5, STATEINF(1)%NUM_ATOMS=22, STATEINF(1)%NUM_STATES=2,
STATEINF(2)%FIRST_ATOM=276, STATEINF(2)%FIRST_CHARGE=139,
STATEINF(2)%FIRST_STATE=7, STATEINF(2)%NUM_ATOMS=16, STATEINF(2)%NUM_STATES=5,
STATEINF(3)%FIRST_ATOM=306, STATEINF(3)%FIRST_CHARGE=219,
STATEINF(3)%FIRST_STATE=12, STATEINF(3)%NUM_ATOMS=21,
STATEINF(3)%NUM_STATES=2, STATEINF(4)%FIRST_ATOM=358,
STATEINF(4)%FIRST_CHARGE=219, STATEINF(4)%FIRST_STATE=12,
STATEINF(4)%NUM_ATOMS=21, STATEINF(4)%NUM_STATES=2,
STATEINF(5)%FIRST_ATOM=500, STATEINF(5)%FIRST_CHARGE=95,
STATEINF(5)%FIRST_STATE=5, STATEINF(5)%NUM_ATOMS=22, STATEINF(5)%NUM_STATES=2,
STATEINF(6)%FIRST_ATOM=542, STATEINF(6)%FIRST_CHARGE=0,
STATEINF(6)%FIRST_STATE=0, STATEINF(6)%NUM_ATOMS=19, STATEINF(6)%NUM_STATES=5,
STATEINF(7)%FIRST_ATOM=741, STATEINF(7)%FIRST_CHARGE=139,
STATEINF(7)%FIRST_STATE=7, STATEINF(7)%NUM_ATOMS=16, STATEINF(7)%NUM_STATES=5,
STATEINF(8)%FIRST_ATOM=789, STATEINF(8)%FIRST_CHARGE=139,
STATEINF(8)%FIRST_STATE=7, STATEINF(8)%NUM_ATOMS=16, STATEINF(8)%NUM_STATES=5,
STATEINF(9)%FIRST_ATOM=805, STATEINF(9)%FIRST_CHARGE=219,
STATEINF(9)%FIRST_STATE=12, STATEINF(9)%NUM_ATOMS=21,
STATEINF(9)%NUM_STATES=2, STATEINF(10)%FIRST_ATOM=1028,
STATEINF(10)%FIRST_CHARGE=139, STATEINF(10)%FIRST_STATE=7,
STATEINF(10)%NUM_ATOMS=16, STATEINF(10)%NUM_STATES=5,
STATEINF(11)%FIRST_ATOM=1326, STATEINF(11)%FIRST_CHARGE=139,
STATEINF(11)%FIRST_STATE=7, STATEINF(11)%NUM_ATOMS=16,
STATEINF(11)%NUM_STATES=5, STATEINF(12)%FIRST_ATOM=1446,
STATEINF(12)%FIRST_CHARGE=95, STATEINF(12)%FIRST_STATE=5,
STATEINF(12)%NUM_ATOMS=22, STATEINF(12)%NUM_STATES=2,
STATEINF(13)%FIRST_ATOM=1468, STATEINF(13)%FIRST_CHARGE=95,
STATEINF(13)%FIRST_STATE=5, STATEINF(13)%NUM_ATOMS=22,
STATEINF(13)%NUM_STATES=2, STATEINF(14)%FIRST_ATOM=1536,
STATEINF(14)%FIRST_CHARGE=139, STATEINF(14)%FIRST_STATE=7,
STATEINF(14)%NUM_ATOMS=16, STATEINF(14)%NUM_STATES=5,
STATEINF(15)%FIRST_ATOM=1767, STATEINF(15)%FIRST_CHARGE=95,
STATEINF(15)%FIRST_STATE=5, STATEINF(15)%NUM_ATOMS=22,
STATEINF(15)%NUM_STATES=2, STATEINF(16)%FIRST_ATOM=1810,
STATEINF(16)%FIRST_CHARGE=139, STATEINF(16)%FIRST_STATE=7,
STATEINF(16)%NUM_ATOMS=16, STATEINF(16)%NUM_STATES=5,
STATENE=0,14.4542090223,14.4542090223,14.4542090223,14.4542090223,
-0.946105574619,0,0,32.3880313021,32.3880313021,32.3880313021,32.3880313021,0,
-78.310003685,
TRESCNT=17,
/https://github.com/choderalab/openmm-constph
Openmm-ConstpH
Amber files are read into openMM using a MonteCarloTitration driver object
# Parameters.
niterations = 500 # number of dynamics/titration cycles to run
nsteps = 500 # number of timesteps of dynamics per iteration
temperature = 300.0 * units.kelvin
timestep = 1.0 * units.femtoseconds
collision_rate = 9.1 / units.picoseconds
pH = 7.0
# Filenames.
prmtop_filename = 'amber-example/prmtop'
inpcrd_filename = 'amber-example/min.x'
cpin_filename = 'amber-example/cpin'
# Load the AMBER system.
import simtk.openmm.app as app
inpcrd = app.AmberInpcrdFile(inpcrd_filename)
prmtop = app.AmberPrmtopFile(prmtop_filename)
system = prmtop.createSystem(implicitSolvent=app.OBC2, nonbondedMethod=app.NoCutoff, constraints=app.HBonds)
# Instantiate system driver
mc_titration = MonteCarloTitration(system, temperature, pH, prmtop, cpin_filename, debug=True)
# Create integrator and context.
platform_name = 'OpenCL'
platform = openmm.Platform.getPlatformByName(platform_name)
integrator = openmm.LangevinIntegrator(temperature, collision_rate, timestep)
context = openmm.Context(system, integrator, platform)
context.setPositions(inpcrd.getPositions())
# Etc. etc.https://github.com/choderalab/openmm-constph
MonteCarloTitration
Current structure of the MonteCarloTitration driver contains methods such as
def addTitratableGroup(self, atom_indices):
"""
Define a new titratable group.
ARGUMENTS
atom_indices (list of int) - the atom indices defining the titration group
"""
def addTitrationState(self, titration_group_index, pKref, relative_energy, charges, proton_count):
"""
Add a titration state to a titratable group.
"""
def update(self, context):
"""
Perform a Monte Carlo update of the titration state.
"""Future plans
How to improve OpenMM constantpH
#pHallthetime
Future plans: ConstpH
Get rid of the CPIN file in favor of forcefield XML flavored format
<TitratableResidues>
<TitratableResidue name="ASP">
<TitrationState index=0>
<NonbondedForce coulomb14scale="0.833333" lj14scale="0.5">
<Atom type="ASP-OD1" charge="-0.45" sigma="xxxxx" epsilon="xxxxxx"/>
<Atom type="ASP-OD2" charge="-0.288" sigma="xxxxx" epsilon="xxxxxx"/>
<Atom type="ASP-HD2" charge="0.408" sigma="xxxxx" epsilon="xxxxxx"/>
<Atom type="ASP-CG" charge="0.33" sigma="xxxxx" epsilon="xxxxxx"/>
</NonbondedForce>
</TitrationState>
<TitrationState index=1>
<NonbondedForce coulomb14scale="0.833333" lj14scale="0.5">
<Atom type="ASP-OD1" charge="-0.635" sigma="xxxxx" epsilon="xxxxxx"/>
<Atom type="ASP-OD2" charge="-0.635" sigma="xxxxx" epsilon="xxxxxx"/>
<!-- Prevent divide by zero! -->
<Atom type="ASP-HD2" charge="-0.0" sigma="0.000" epsilon="0.000"/>
<Atom type="ASP-CG" charge="0.27" sigma="xxxxx" epsilon="xxxxxx"/>
</NonbondedForce>
</TitrationState>
</TitratableResidue>
</TitratableResidues>#pHallthetime
- Expand to incorporate LJ parameters
- Currently, just charges are supported
- Better support for ligands
- Explicit solvent using NCMC & counter ions
- Inserting ions is unlikely to be accepted because of close contacts, water rearrangement, and more...
- Coordinated changes (pairs of residues, etc.) to increase acceptance probability
- Pick pairs at random
- Find a way to pick pairs close together that satisfies detailed balance
Future plans: ConstpH
- Benchmark pKa calculation tools to find the most reliable reference values for ligands
- Investigate the need for charge corrections in PME calculations (since we are modifying system charges)
- Investigate the need to go into the OpenMM C++/Cuda layer for more efficient calculations
Future plans: ConstpH
#pHallthetime
Systems of interest
- Kinases & FDA approved inhibitors
- Czodrowski et al. protease inhibitors
#pHallthetime
Systems of interest
- Kinases & FDA approved inhibitors
- Czodrowski et al. protease inhibitors
- Lactate dehydrogenase A (LDHA)
#pHallthetime
Kinase inhibitors
pKa predictions using EPIK suggest that many kinase inhibitors have more than one accessible protonation state at pH 7.4
Figure by: John & Julie
Lactate Dehydrogenase A
PDB: 1I10
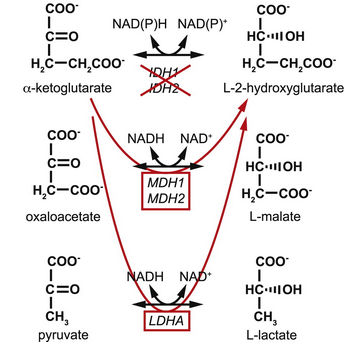
- Discovered a pH dependent effect to the reaction rate
- Continued collaboration with Andrew Intlekofer (Thompson lab)
Intlekofer et al. Cell Metabolism 22, 304–311, August 4, 2015
Lactate Dehydrogenase A
PDB: 1I10
Lactate Dehydrogenase A
PDB: 1I10