End-To-End Testing Pipeline for Phasing and Imputation
1 Factory Pattern
1 Factory Pattern
def main(order):
piper = PiperFactory.create_piper(order)
play_pipe(piper)
def play_pipe(piper):
piper.pipe_stdin()
cmd = piper.render_user_cmd(order=piper.order,)
piper.run_user_script(cmd)
piper.collect_output_files()
piper.upload_output_files()
piper.teardown()1.1
├── seqpiper
│ ├── base
│ ├── bam
│ ├── vcf
│ │ ├── base.py
│ │
│ ├── main.py
│
├── tests
├── phasing.sh
├── imputation.sh
1.2 Advantage of doing this
class VcfPiperBase(PiperAbstractFactory):
def pipe_stdin(self):
some_vcf_function()class PiperAbstractFactory(object):
@abstractmethod
def stdin(self):
pass
def render_user_cmd(self):
some code
def run_user_script(self):
some code
def collect_output_files(self):
some code
def upload_output_files(self):
some code
def teardown(self):
some code2 Prevent Import Error
import sys
import traceback
sys.path.append('/usr/local/seqslab/SeqPiper')
from script.library.GatkWrapper import ApplyRecalibration
from script.common.base import SeqPiperBase
from script.common.order import JobOrder
from script.common.exception import SeqPiperEmptyVariantFile2.2 Formatter Would Do This...
import sys
import traceback
from script.library.GatkWrapper import ApplyRecalibration
from script.common.base import SeqPiperBase
from script.common.order import JobOrder
from script.common.exception import SeqPiperEmptyVariantFile
sys.path.append('/usr/local/seqslab/SeqPiper')2.3 ModuleNotFoundError!

2.4 Build SeqPiper Package
setup(
name="seqpiper",
version="0.0.1",
long_description=readme,
long_description_content_type="text/markdown",
url="",
author="",
author_email="",
license="",
install_requires=[],
keywords="bioinformatics, seqpiper",
packages=find_packages(),
)python3 setup.py install2.5 Remove sys.path.append
import sys
import traceback
from script.library.GatkWrapper import ApplyRecalibration
from script.common.base import SeqPiperBase
from script.common.order import JobOrder
from script.common.exception import SeqPiperEmptyVariantFile3. CI Pipeline
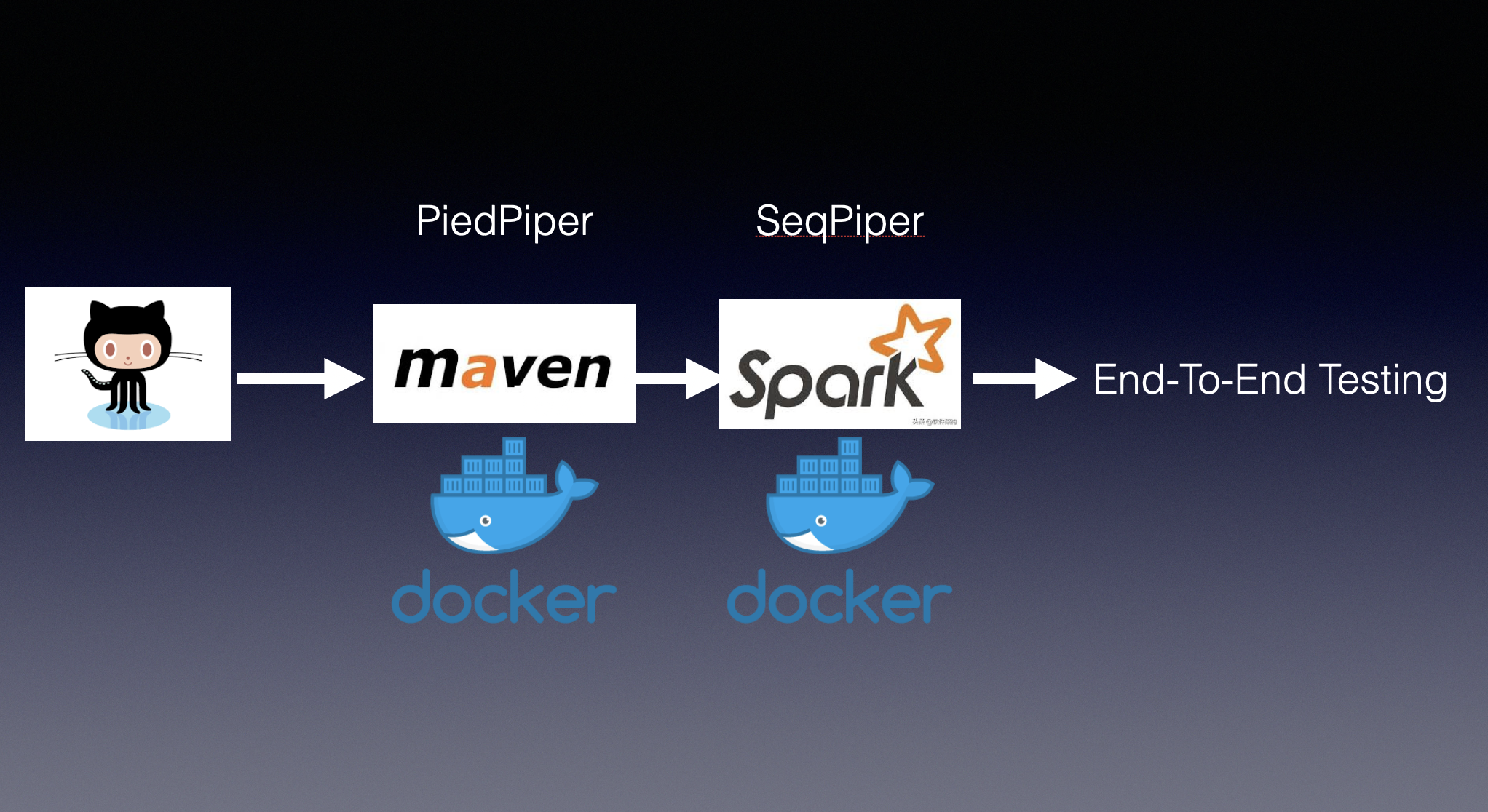
3.1 How to add new test case?
/opt/bitnami/spark/bin/spark-submit \
--class net.vartotal.piper.cli.PiperMain \
--driver-cores 1 \
--driver-memory 1g \
--executor-cores 1 \
--executor-memory 10g \
--conf "spark.dynamicAllocation.enabled=false" \
--conf "spark.executor.extraJavaOptions=-XX:+UseG1GC" \
--conf "spark.serializer=org.apache.spark.serializer.KryoSerializer" \
--conf "spark.kryo.registrator=net.vartotal.piper.serialization.ADAMKryoRegistrator" \
--conf "spark.executor.memoryOverhead=14g" \
--conf "spark.speculation=false" \
/usr/local/seqslab/SeqPiper/target/piper-2.1.0.jar vcf \
--reference-version 38 \
--reference-system GRCH \
--piper-script fixtures.scripts.no_stdout \
-i 0=/usr/local/seqslab/SeqPiper/fixtures/vcf/ \
--samplePath /usr/local/seqslab/SeqPiper/fixtures/vcf/ \
--vcf-output-path vcf_gz_folder \
--partition-bed-path /usr/local/seqslab/SeqPiper/fixtures/bed/38/chromosomes3.2 How to add new test case?
├── seqpiper
│ ├── base
│ ├── bam
│ ├── vcf
│ │ ├── base.py
│ │
│ ├── main.py
│
├── tests
├── phasing.sh
├── imputation.sh
3.3 CI Pipeline
3.4 Advantage of Doing This
- Spend less on AzureBatch for testing
- Save some time to test pipeline