Greene Lab Journal Club
9:00 AM on January 13, 2017

From the 2017 NAR Database Issue

Predicted protein associations from
- systematic co-expression analysis
- detection of shared selective signals across genomes
- automated text-mining of the scientific literature
- computational transfer of interaction knowledge between organisms based on gene orthology
Experimental protein associations from
- experimental data on protein–protein interactions
- importing known pathways and protein complexes from curated databases
- Edge scores for the "likelihood that a given interaction is biologically meaningful, specific and reproducible, given the supporting evidence."
- Version 10.5 revamped the frontend
- Online at http://string-db.org/
- Similar to imp.princeton.edu?
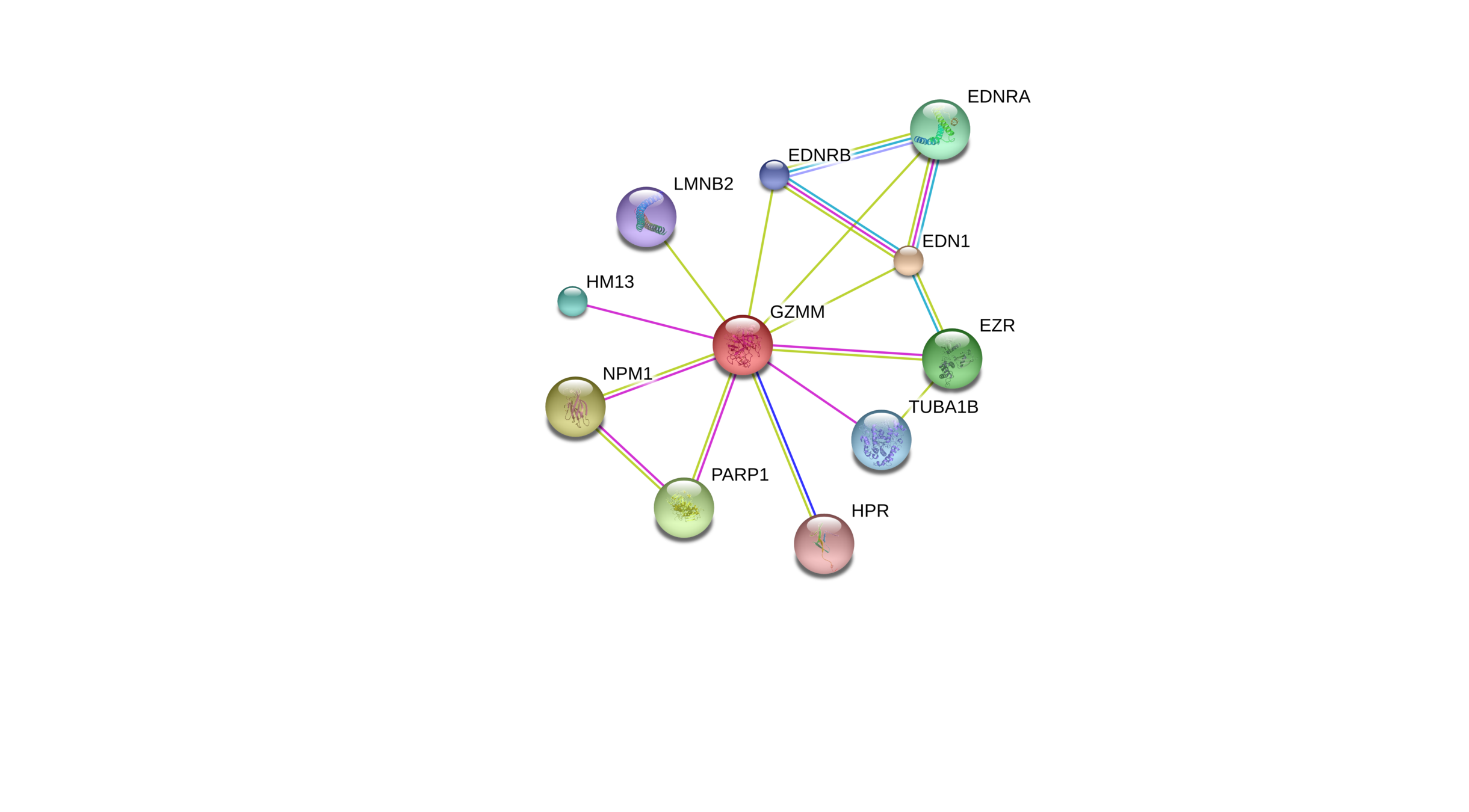
STRING aims to set itself apart in three ways:
-
comprehensiveness – it covers the largest number of organisms and uses the widest breadth of input sources, including automated text-mining and computational predictions,
-
usability – in terms of an intuitive web interface, Cytoscape integration and programmatic access options, and
- quality control and traceability – each interaction is annotated with benchmarked confidence scores, separately per evidence type, and the underlying evidence can be tracked to its source. STRING has been maintained continuously since the year 2000.

As of September 2016, we have 39,870 active entries within our HGNC database of which 19,017 are for protein-coding genes.
Over the last three years:
- total number of protein-coding genes has been relatively stable
- over 200 entries were reclassified to or from the protein-coding locus type as new evidence has become available
The majority of HGNC's human gene symbols have become or are becoming well entrenched in the literature and databases, and should never require reassignment.
HGNC limit the number of symbol alterations and only ever change symbols for specific reasons, the most common being if a symbol can be updated from an uninformative placeholder designation, such as our C$orf#s, KIAA# or FAM#s, or if the symbol was originally assigned based on information that has since been found to be erroneous and the existing symbol could be misleading. To avoid symbol changes but improve our nomenclature we can make updates to gene names while retaining the symbols, for example, to remove references to other species or human-specific phenotypes.
Motivation: I recently used a hack to group genes.
Genes are grouped into families based on homology or a shared characteristic such as a common function and/or phenotype, or membership of a complex. We have also begun curating the relationships between different gene families, for example, the protein phosphatase gene family members represent a subset of the phosphatases gene family.
See also https://doi.org/bw6j
Gene family reports often feature a description of the family, which may be sourced from Wikipedia, UniProt, a publication, or may be written by an HGNC curator or specialist advisor.

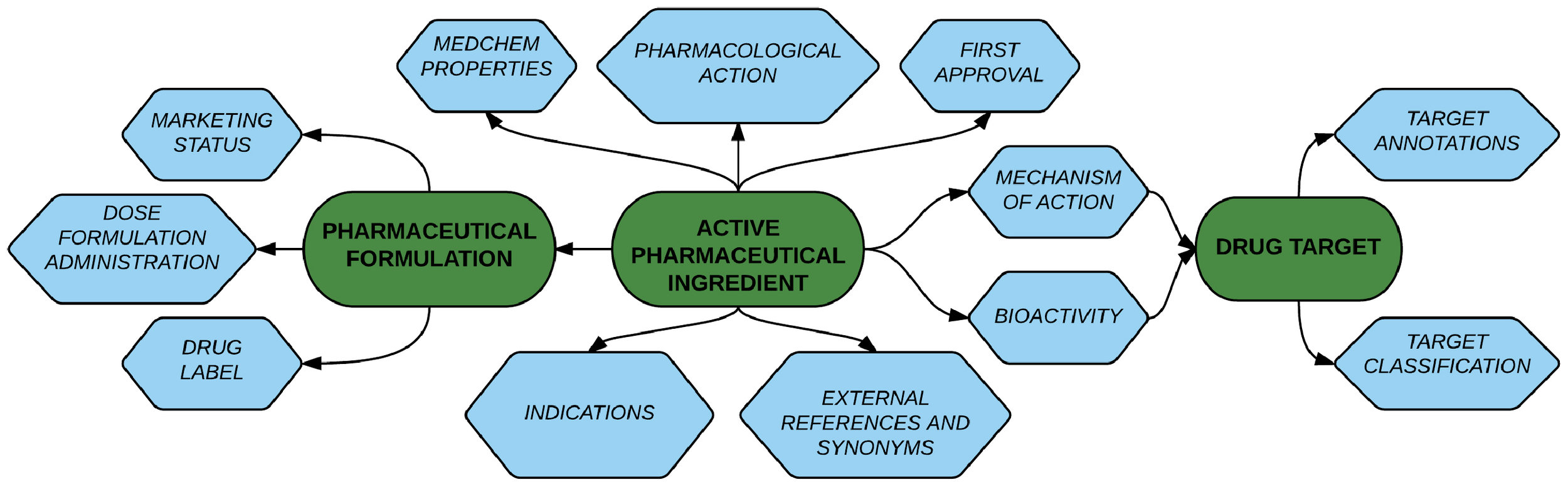
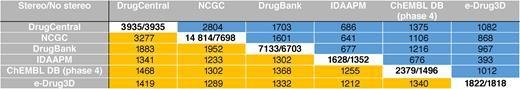
Database licensed as CC BY-SA, drugcentral.org

| Aspect | Terms (classes) | Relationships |
|---|---|---|
| Molecular function (MF) | 10,417 | 14,039 |
| Cellular component (CC) | 4,022 | 7,854 |
| Biological process (BP) | 29,146 | 71,372 |
The GO describes function with respect to three aspects:
- molecular function (molecular-level activities performed by gene products),
- cellular component (the locations relative to cellular structures in which a gene product performs a function),
- biological process (the larger processes, or ‘biological programs’ accomplished by multiple molecular activities).
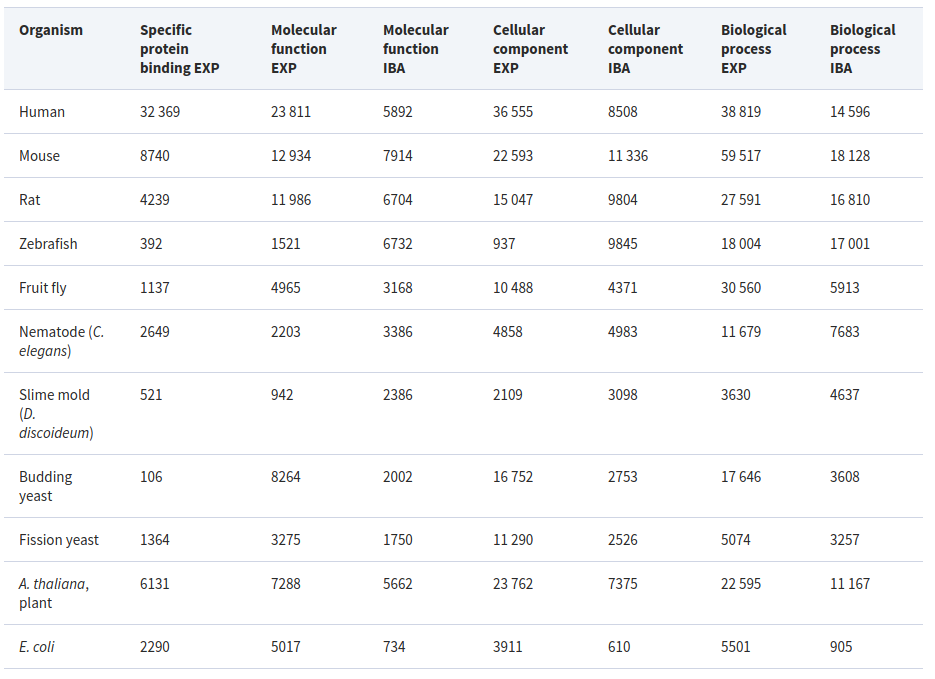
Experimental (EXP) and phylogenetically inferred (IBA) annotations
Annotations with direct experimental evidence are created by biocurators, PhD-level experts trained in computational knowledge representation, who read peer-reviewed literature and create GO annotations as justified by the evidence presented in those articles.

