Greene Lab Journal Club
2018-01-09
https://doi.org/cgwt
Why L1000 Platform?
- the high cost of commercial gene expression microarrays and even RNA sequencing (RNA-seq) precludes such a genome-scale CMap.
- The reagent cost of the L1000 assay is approximately two dollars.
- The L1000 platform is hybridization based, thus making the detection of non-abundant transcripts feasible.
Scope of resource
In total, we generated 1,319,138 L1000 profiles from 42,080 perturbagens (
- 19,811 small molecule compounds,
- 18,493 shRNAs,
- 3,462 cDNAs,
- and 314 biologics),
corresponding to 25,200 biological entities (
- 19,811 compounds,
- shRNA and/or cDNA against 5,075 genes,
- and 314 biologics)
for a total of 473,647 signatures (consolidating replicates)
Which Genes
- We hypothesized that it might be possible to capture at low cost any cellular state by measuring a reduced representation of the transcriptome. To explore this, we analyzed 12,031 Affymetrix HGU133A expression profiles in the Gene Expression Omnibus (GEO).
- This analysis showed that 1,000 landmarks were sufficient to recover 82% of the information in the full transcriptome
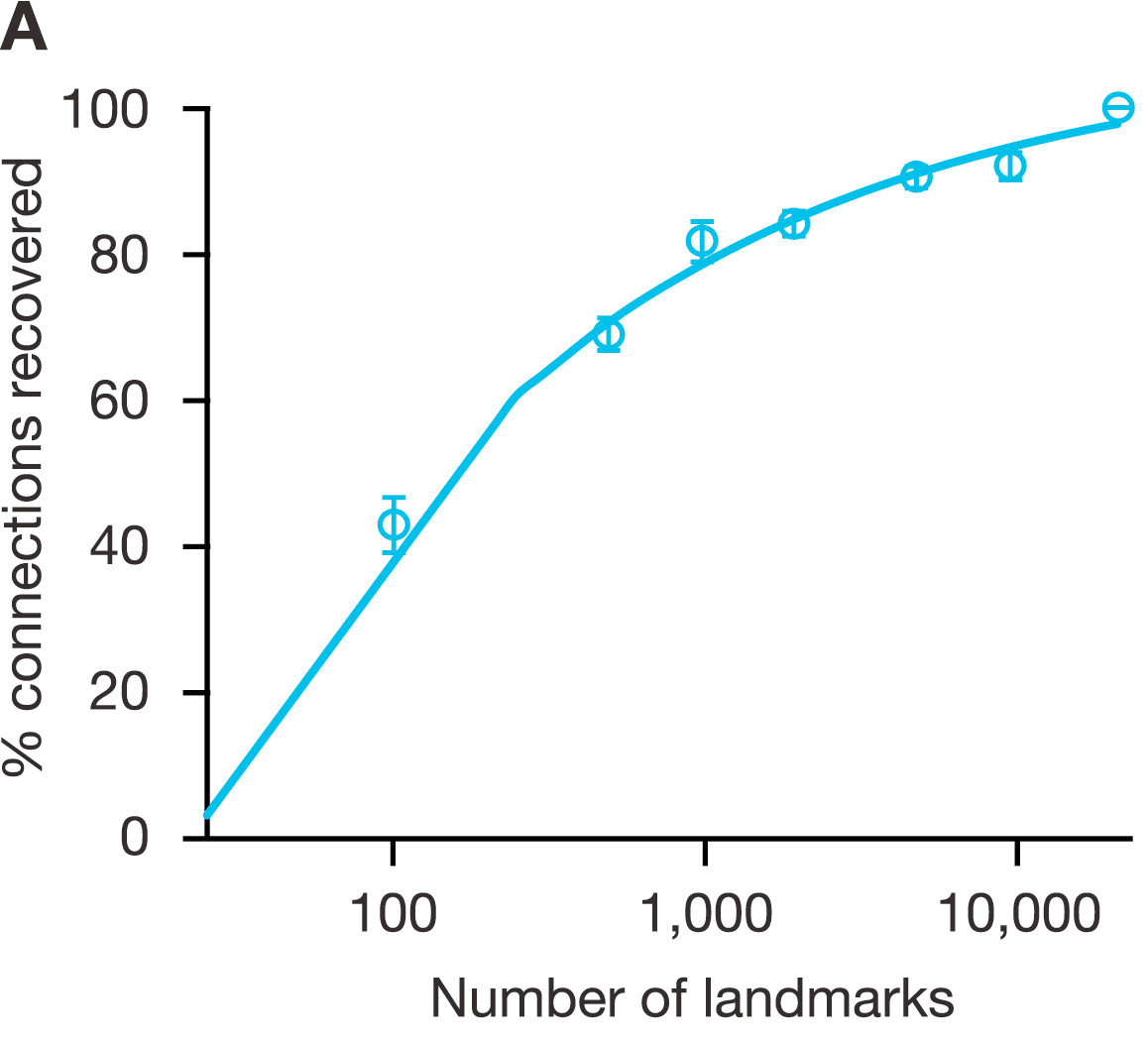
Figure S1A: Evaluating dimensionality reduction. Simulation showing the mean percentage (+/− SEM) of 33 benchmark connections recovered from an inferred CMap pilot dataset as a function of number of landmark genes used to train the inference model, indicating that around 1000 landmarks were sufficient to recover 82% of expected connections
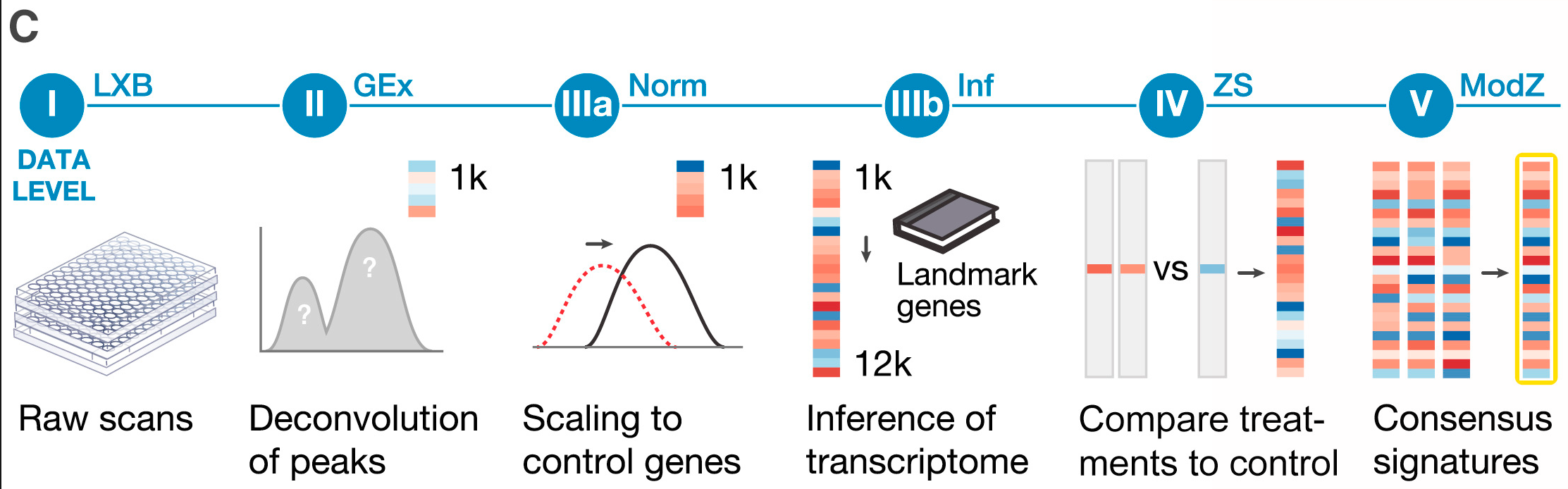
Figure 2C: Signature generation and data levels.
(I) Raw bead count and fluorescence intensity measured by Luminex scanners.
(II) Deconvoluted data to assign expression levels to two transcripts measured on the same bead.
(IIIa) Normalization to adjust for non-biological variation.
(IIIb) Inferred expression of 12,328 genes from measurement of 978 landmarks.
(IV) Differential expression values.
(V) Signatures representing collapse of replicate profiles.
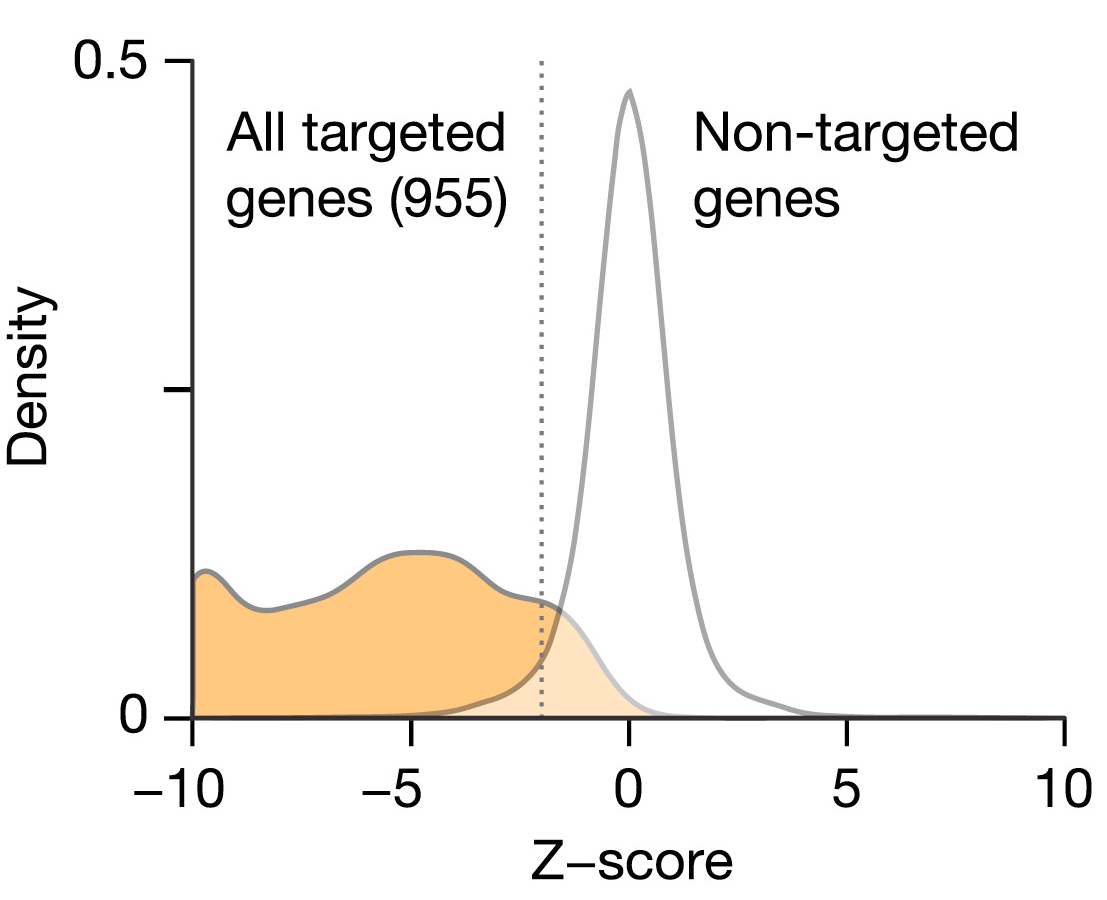
Figure 2C: Validation of L1000 probes using shRNA knockdown. MCF7 and PC3 cells transduced with shRNAs targeting 955 landmark genes. Differential expression values (z-scores) were computed for each landmark, and the percentile rank of expression z-scores in the experiment in which it was targeted relative to all other experiments was computed. 841/955 genes (88%) rank in the top 1% of all experiments and 907/955 (95%) rank in the top 5%. ... Middle panel: z-score distribution from all targeted (orange) and non-targeted (white) genes. Distribution from the targeted set is significantly lower than non-targeted (p value < 10−16).
See however https://github.com/dhimmel/lincs/issues/4: imputed target genes are not differentially-expressed in the expected direction
As CSNK1A1 was among the 3,799 genes subjected to shRNA-mediated knockdown, we used the CMap to generate a signature of CSNK1A1 loss of function (LoF). We then queried all compounds in the database against this signature to identify perturbations that phenocopied CSNK1A1 loss. One unannotated compound, BRD-1868, showed strong connectivity to CSNK1A1 knockdown in two cell types. This suggested that BRD-1868 might function as a novel CSNK1A1 inhibitor.
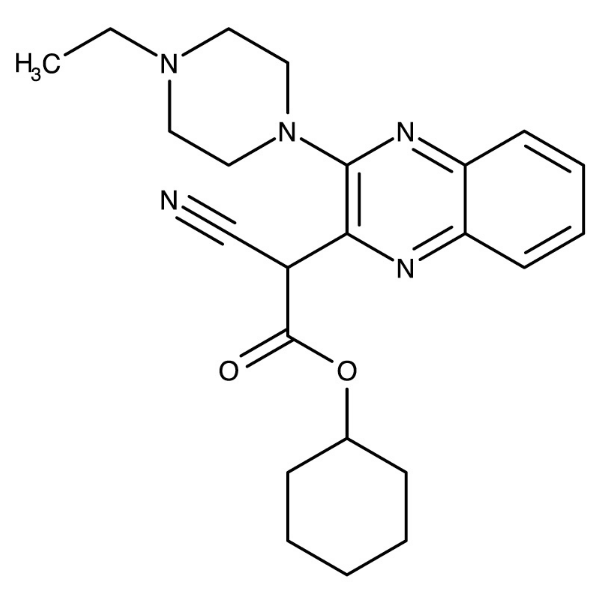
Application: finding a CSNK1A1 inhibitor
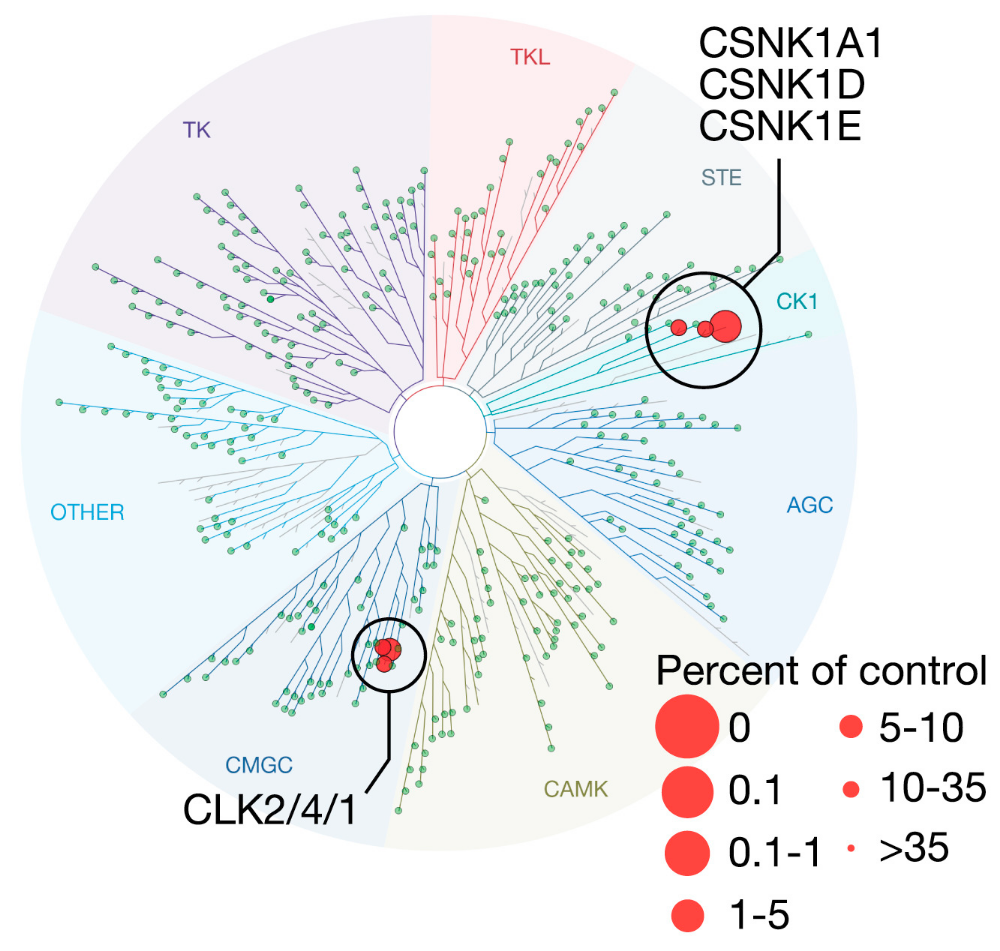
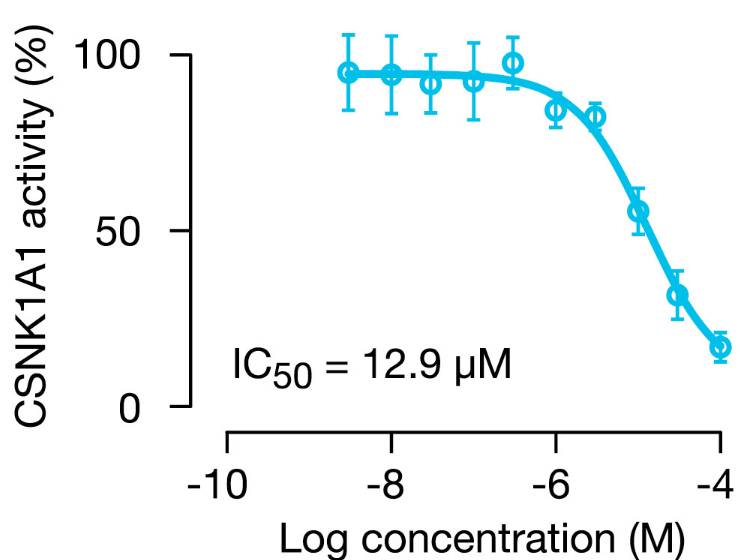
Figure 6B: Discovery of novel CSNK1A1 inhibitor.
- TREEspot image of Kinomescan binding assay performed with BRD-1868 at 10 μM demonstrated inhibition of 6/456 kinases tested, including CSNK1A1.
- BRD-1868 inhibits phosphorylation of peptide substrate by CSNK1A1, with IC50 12.9 μM. Error bars indicate standard deviation between technical replicates.


https://doi.org/ch4b
Quotes
- Recently, there has been an expanding focus on the microbial ecology of the “built environment”—human constructed entities like buildings, cars, and trains—places where humans spend a large fraction of their time. One relatively unexplored type of built environment is that found in space
- We have also compiled a collection of papers on space microbiology
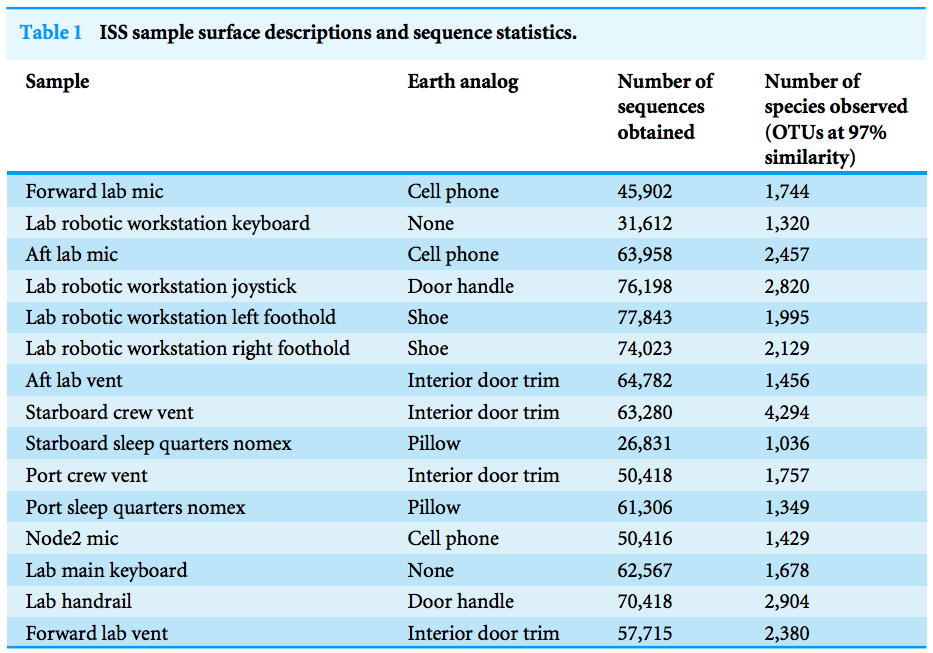
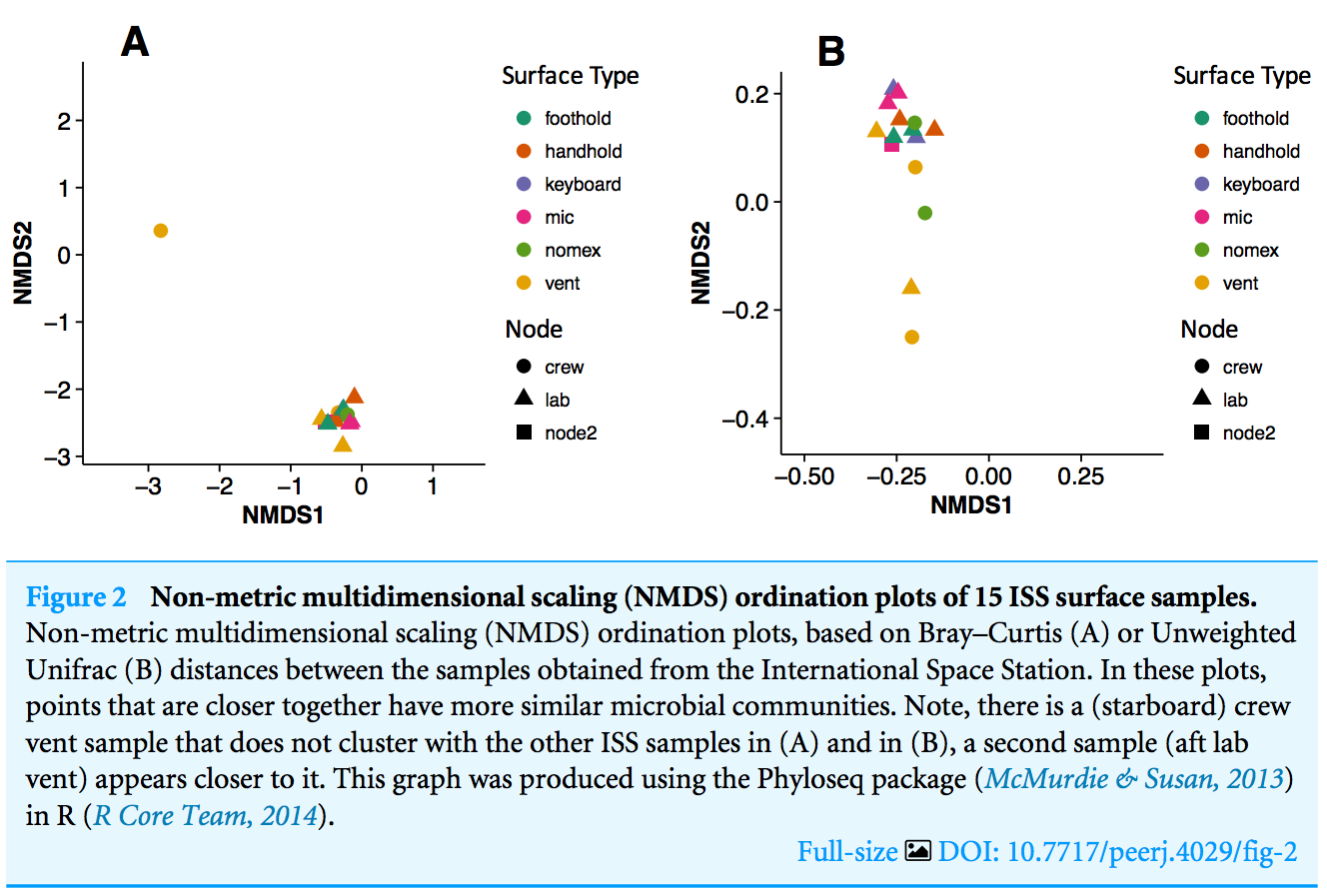
there is one sample, the starboard crew vent, that appears distinct from all of the other samples...
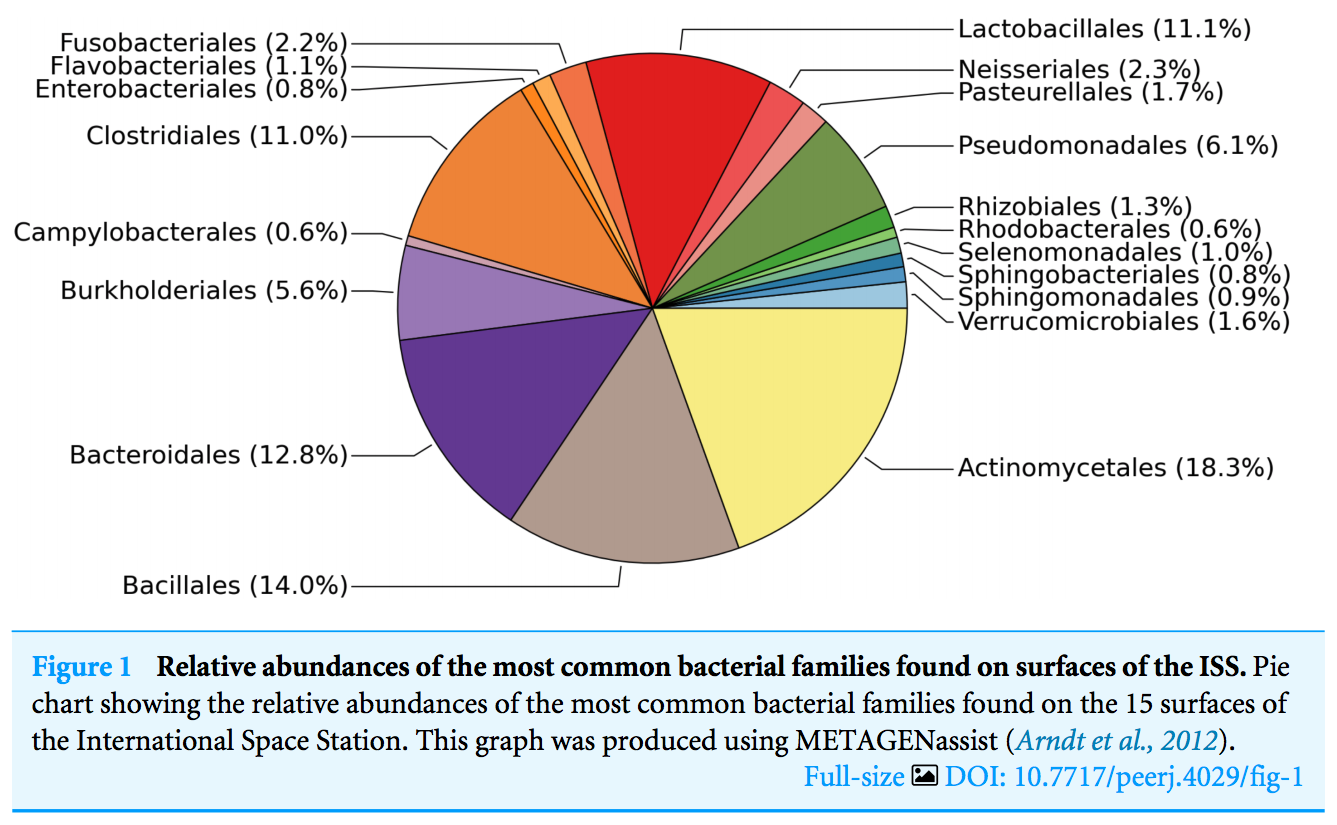
The three most abundant families in the starboard crew vent sample are Bacteroidaceae, Ruminococcaceae, and Verrumicrobiaceae (comprising 60.1% of all sequences)
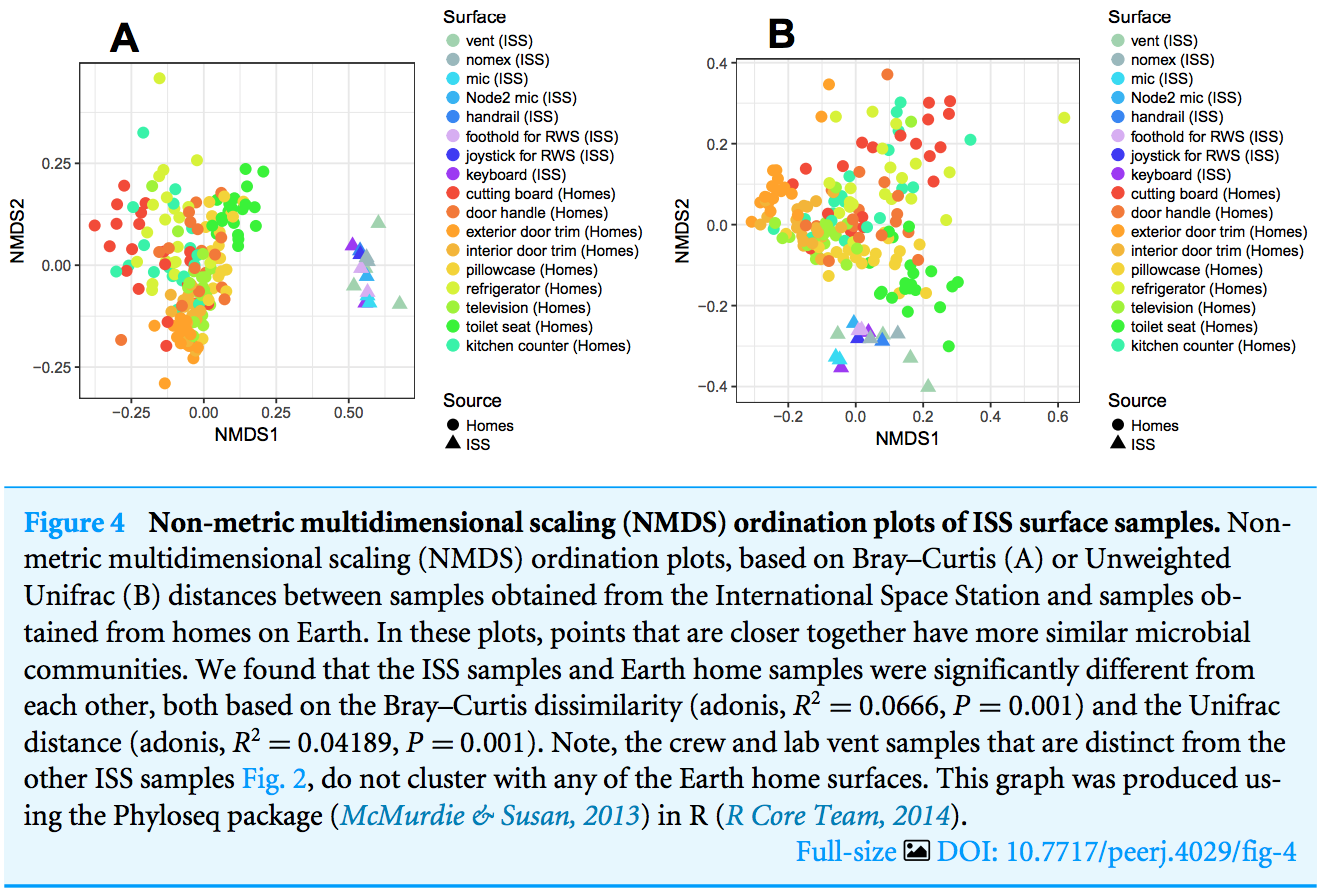
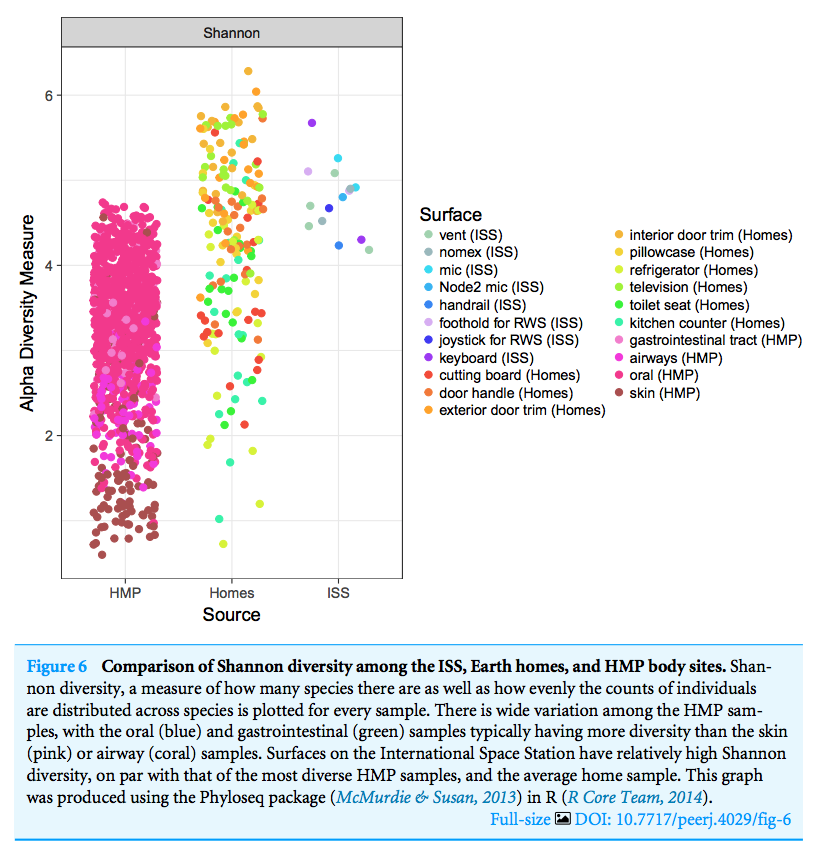
See https://doi.org/ch4r

https://github.com/emreg00/repurpose
The Gist:
- To identify potential candidates that could be repositioned for a new indication, many studies make use of chemical, target, and side effect similarity between drugs to train classifiers.
- their use in practice, such as for rare diseases, is hindered by the assumption that there are already known and similar drugs for a given condition of interest.
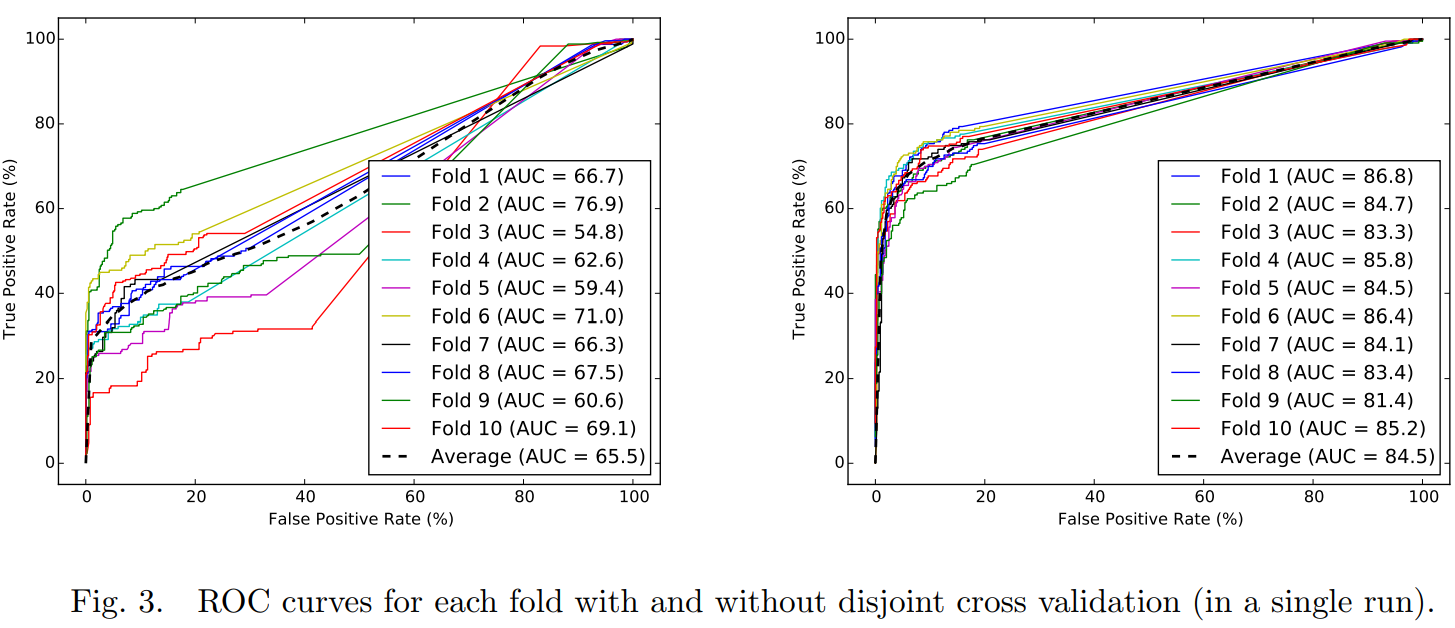
- while a simple chemical, target, and side effect similarity based machine learning method can achieve good performance on the benchmark data set, the prediction performance drops sharply when the drugs in the folds of the cross validation are not overlapping and the similarity information within the training and test sets are used independently.
- need for unsupervised approaches to identify novel drug uses inside the unexplored pharmacological space
Existing studies often assume that the drugs that are in the test set will also appear in the training set, a rather counter-intuitive assumption as, in practice, one is often interested in predicting truly novel drug-disease associations (i.e. for drugs that have no known indications previously). We challenge this assumption by evaluating the effect of having training and test sets in which none of the drugs in one overlaps with the drugs in the other. Accordingly, we implement a disjoint cross validation fold generation method that ensures that the drug-disease pairs are split such that none of the drugs in the training set appear in the test set

Fig. 4. Prediction accuracy (AUC) when each similarity feature used individually in disjoint cross validation. Error bars show standard deviation of AUC over ten runs of ten-fold cross validation.

Luo et al. use an independent set of drug-disease associations, yet, 95% of the drugs in the independent set are also in the original data set (109 out of 115).
On the other hand, Gottlieb et al. create the folds such that 10% of the drugs are hidden instead of 10% of the drug-disease pairs, but they do not ensure that the drugs used to train the model are disjoint from the drugs in the test set.
Previous Approaches
See https://doi.org/ch44

Proceedings of the VLDB Endowment, November 2017
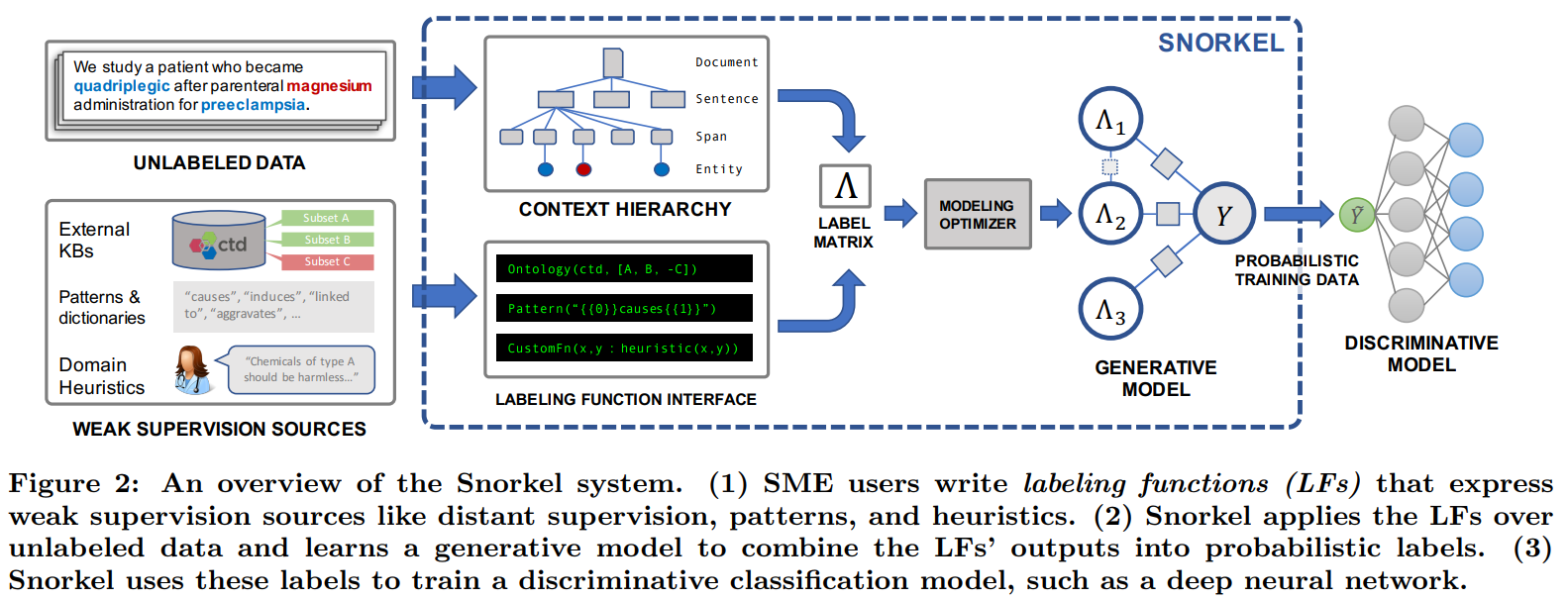
Snorkel Architecture
- Writing Labeling Functions: Rather than hand-labeling training data, users of Snorkel write labeling functions, which allow them to express various weak supervision sources such as patterns, heuristics, external knowledge bases, and more. This was the component most informed by early interactions (and mistakes) with users over the last year of deployment, and we present a flexible interface and supporting data model.
- Modeling Accuracies and Correlations: Next, Snorkel automatically learns a generative model over the labeling functions, which allows it to estimate their accuracies and correlations. This step uses no ground-truth data, learning instead from the agreements and disagreements of the labeling functions. We observe that this step improves end predictive performance 5.81% over Snorkel with unweighted label combination, and anecdotally that it streamlines the user development experience by providing actionable feedback about labeling function quality.
- Training a Discriminative Model: The output of Snorkel is a set of probabilistic labels that can be used to train a wide variety of state-of-the-art machine learning models, such as popular deep learning models. While the generative model is essentially a re-weighted combination of the user-provided labeling functions—which tend to be precise but low-coverage—modern discriminative models can retain this precision while learning to generalize beyond the labeling functions, increasing coverage and robustness on unseen data.
Declarative Labeling Functions: Snorkel includes a library of declarative operators that encode the most common weak supervision function types, based on our experience with users over the last year. These functions capture a range of common forms of weak supervision, for example:
- Pattern-based: Pattern-based heuristics embody the motivation of soliciting higher information density input from SMEs. For example, pattern-based heuristics encompass feature annotations [51] and pattern-bootstrapping approaches [18, 20] (Example 2.3).
- Distant supervision: Distant supervision generates training labels by heuristically aligning data points with an external knowledge base, and is one of the most popular forms of weak supervision [4, 22, 32].
- Weak classifiers: Classifiers that are insucient for our task—e.g., limited coverage, noisy, biased, and/or trained on a different dataset—can be used as labeling functions.
- Labeling function generators: One higher-level abstraction that we can build on top of labeling functions in Snorkel is labeling function generators, which generate multiple labeling functions from a single resource, such as crowdsourced labels and distant supervision from structured knowledge bases (Example 2.4).
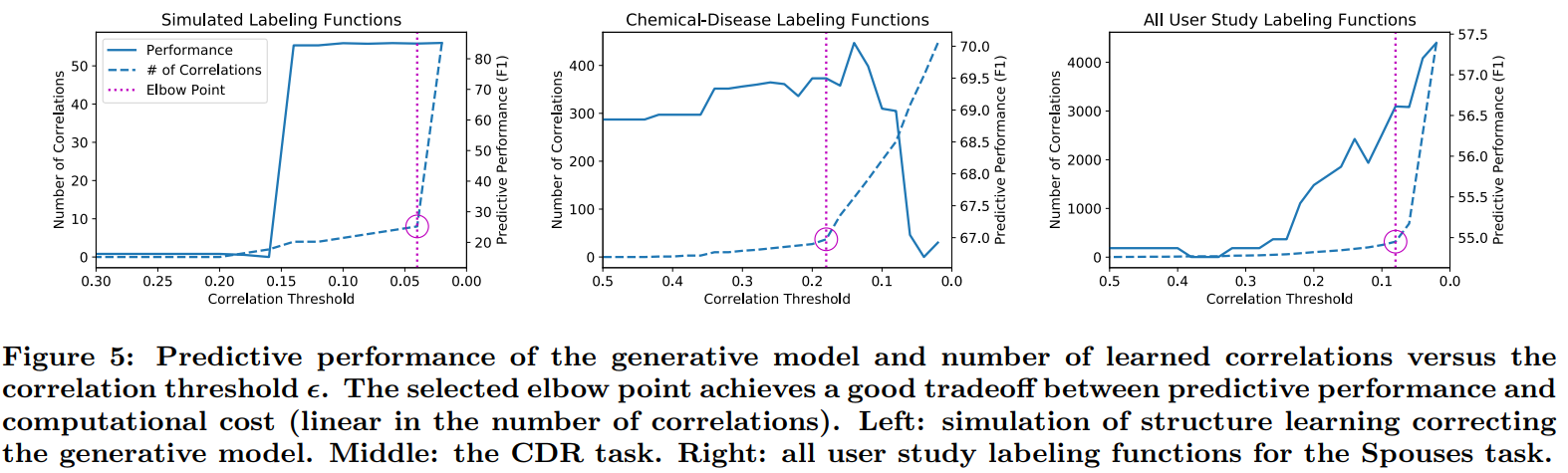
this approach relies on a selection threshold hyperparameter ϵ which induces a tradeoff space between predictive performance and computational cost.