FlowCraft: A modular, extensible and flexible tool to build, monitor and report nextflow pipelines
Diogo N Silva
Workflows in the Paleolithic era
Writing of pipelines in python/perl/shell scripts circa 2000, colorized.

- Custom ad-hoc scripts
- Difficult to parallelise
- Difficult to install/run
- Hard to deploy in multiple environments
- What's workflow managers?!
- What's docker?!
Workflows in the Modern era
The game changing combination of workflow managers + containers:
- Portability
- Reproducible
- Scalability
- Multi-scale containerization
- Native cloud support

Workflows in the Modern era
Challenges persist
- Fast pace of bioinformatics software landscape
- Continuous need for benchmarking and comparative analyses
- The need for agile and dynamic pipeline building
- Remove the pain of changing inner workings of workflows
>3000 bioinformatics tools developed in 2018
Pipelines need to be disposable
Pipelines, like tools, are temporary
The lingering problem
#!/usr/bin/env nextflow
/*
========================================================================================
nf-core/chipseq
========================================================================================
nf-core/chipseq Analysis Pipeline.
#### Homepage / Documentation
https://github.com/nf-core/chipseq
----------------------------------------------------------------------------------------
*/
def helpMessage() {
log.info nfcoreHeader()
log.info"""
Usage:
The typical command for running the pipeline is as follows:
nextflow run nf-core/chipseq --design design.csv --genome GRCh37 -profile docker
Mandatory arguments:
--design Comma-separated file containing information about the samples in the experiment (see docs/usage.md)
--fasta Path to Fasta reference. Not mandatory when using reference in iGenomes config via --genome
--gtf Path to GTF file in Ensembl format. Not mandatory when using reference in iGenomes config via --genome
-profile Configuration profile to use. Can use multiple (comma separated)
Available: conda, docker, singularity, awsbatch, test
Generic
--singleEnd Specifies that the input is single-end reads
--seq_center Sequencing center information to be added to read group of BAM files
--fragment_size [int] Estimated fragment size used to extend single-end reads (Default: 200)
--fingerprint_bins [int] Number of genomic bins to use when calculating fingerprint plot (Default: 500000)
References If not specified in the configuration file or you wish to overwrite any of the references
--genome Name of iGenomes reference
--bwa_index Full path to directory containing BWA index including base name i.e. /path/to/index/genome.fa
--gene_bed Path to BED file containing gene intervals
--tss_bed Path to BED file containing transcription start sites
--macs_gsize Effective genome size parameter required by MACS2. If using iGenomes config, values have only been provided when --genome is set as GRCh37, GRCm38, hg19, mm10, BDGP6 and WBcel235
--blacklist Path to blacklist regions (.BED format), used for filtering alignments
--saveGenomeIndex If generated by the pipeline save the BWA index in the results directory
Trimming
--clip_r1 [int] Instructs Trim Galore to remove bp from the 5' end of read 1 (or single-end reads) (Default: 0)
--clip_r2 [int] Instructs Trim Galore to remove bp from the 5' end of read 2 (paired-end reads only) (Default: 0)
--three_prime_clip_r1 [int] Instructs Trim Galore to remove bp from the 3' end of read 1 AFTER adapter/quality trimming has been performed (Default: 0)
--three_prime_clip_r2 [int] Instructs Trim Galore to re move bp from the 3' end of read 2 AFTER adapter/quality trimming has been performed (Default: 0)
--skipTrimming Skip the adapter trimming step
--saveTrimmed Save the trimmed FastQ files in the the results directory
Alignments
--keepDups Duplicate reads are not filtered from alignments
--keepMultiMap Reads mapping to multiple locations are not filtered from alignments
--saveAlignedIntermediates Save the intermediate BAM files from the alignment step - not done by default
Peaks
--narrowPeak Run MACS2 in narrowPeak mode
--broad_cutoff [float] Specifies broad cutoff value for MACS2. Only used when --narrowPeak isnt specified (Default: 0.1)
--min_reps_consensus Number of biological replicates required from a given condition for a peak to contribute to a consensus peak (Default: 1)
--saveMACSPileup Instruct MACS2 to create bedGraph files normalised to signal per million reads
--skipDiffAnalysis Skip differential binding analysis
QC
--skipFastQC Skip FastQC
--skipPicardMetrics Skip Picard CollectMultipleMetrics
--skipPreseq Skip Preseq
--skipPlotProfile Skip deepTools plotProfile
--skipPlotFingerprint Skip deepTools plotFingerprint
--skipSpp Skip Phantompeakqualtools
--skipIGV Skip IGV
--skipMultiQC Skip MultiQC
--skipMultiQCStats Exclude general statistics table from MultiQC report
Other
--outdir The output directory where the results will be saved
--email Set this parameter to your e-mail address to get a summary e-mail with details of the run sent to you when the workflow exits
--maxMultiqcEmailFileSize Theshold size for MultiQC report to be attached in notification email. If file generated by pipeline exceeds the threshold, it will not be attached (Default: 25MB)
-name Name for the pipeline run. If not specified, Nextflow will automatically generate a random mnemonic
AWSBatch
--awsqueue The AWSBatch JobQueue that needs to be set when running on AWSBatch
--awsregion The AWS Region for your AWS Batch job to run on
""".stripIndent()
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- SET UP CONFIGURATION VARIABLES -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* SET UP CONFIGURATION VARIABLES
*/
// Show help message
if (params.help){
helpMessage()
exit 0
}
// Check if genome exists in the config file
if (params.genomes && params.genome && !params.genomes.containsKey(params.genome)) {
exit 1, "The provided genome '${params.genome}' is not available in the iGenomes file. Currently the available genomes are ${params.genomes.keySet().join(", ")}"
}
////////////////////////////////////////////////////
/* -- DEFAULT PARAMETER VALUES -- */
////////////////////////////////////////////////////
// Configurable variables
params.fasta = params.genome ? params.genomes[ params.genome ].fasta ?: false : false
params.bwa_index = params.genome ? params.genomes[ params.genome ].bwa ?: false : false
params.gtf = params.genome ? params.genomes[ params.genome ].gtf ?: false : false
params.gene_bed = params.genome ? params.genomes[ params.genome ].gene_bed ?: false : false
params.macs_gsize = params.genome ? params.genomes[ params.genome ].macs_gsize ?: false : false
params.blacklist = params.genome ? params.genomes[ params.genome ].blacklist ?: false : false
// Has the run name been specified by the user?
// this has the bonus effect of catching both -name and --name
custom_runName = params.name
if (!(workflow.runName ==~ /[a-z]+_[a-z]+/)){
custom_runName = workflow.runName
}
////////////////////////////////////////////////////
/* -- CONFIG FILES -- */
////////////////////////////////////////////////////
// Pipeline config
ch_output_docs = file("$baseDir/docs/output.md", checkIfExists: true)
// JSON files required by BAMTools for alignment filtering
if (params.singleEnd) {
ch_bamtools_filter_config = file(params.bamtools_filter_se_config, checkIfExists: true)
} else {
ch_bamtools_filter_config = file(params.bamtools_filter_pe_config, checkIfExists: true)
}
// Header files for MultiQC
ch_multiqc_config = file(params.multiqc_config, checkIfExists: true)
ch_peak_count_header = file("$baseDir/assets/multiqc/peak_count_header.txt", checkIfExists: true)
ch_frip_score_header = file("$baseDir/assets/multiqc/frip_score_header.txt", checkIfExists: true)
ch_peak_annotation_header = file("$baseDir/assets/multiqc/peak_annotation_header.txt", checkIfExists: true)
ch_deseq2_pca_header = file("$baseDir/assets/multiqc/deseq2_pca_header.txt", checkIfExists: true)
ch_deseq2_clustering_header = file("$baseDir/assets/multiqc/deseq2_clustering_header.txt", checkIfExists: true)
ch_spp_correlation_header = file("$baseDir/assets/multiqc/spp_correlation_header.txt", checkIfExists: true)
ch_spp_nsc_header = file("$baseDir/assets/multiqc/spp_nsc_header.txt", checkIfExists: true)
ch_spp_rsc_header = file("$baseDir/assets/multiqc/spp_rsc_header.txt", checkIfExists: true)
////////////////////////////////////////////////////
/* -- VALIDATE INPUTS -- */
////////////////////////////////////////////////////
// Validate inputs
if (params.design) { ch_design = file(params.design, checkIfExists: true) } else { exit 1, "Samples design file not specified!" }
if (params.gtf) { ch_gtf = file(params.gtf, checkIfExists: true) } else { exit 1, "GTF annotation file not specified!" }
if (params.gene_bed) { ch_gene_bed = file(params.gene.bed, checkIfExists: true) }
if (params.tss_bed) { ch_tss_bed = file(params.tss_bed, checkIfExists: true) }
if (params.blacklist) { ch_blacklist = file(params.blacklist, checkIfExists: true) }
if (params.fasta){
lastPath = params.fasta.lastIndexOf(File.separator)
bwa_base = params.fasta.substring(lastPath+1)
ch_fasta = file(params.fasta, checkIfExists: true)
} else {
exit 1, "Fasta file not specified!"
}
if (params.bwa_index){
lastPath = params.bwa_index.lastIndexOf(File.separator)
bwa_dir = params.bwa_index.substring(0,lastPath+1)
bwa_base = params.bwa_index.substring(lastPath+1)
ch_bwa_index = Channel
.fromPath(bwa_dir, checkIfExists: true)
.ifEmpty { exit 1, "BWA index directory not found: ${bwa_dir}" }
}
////////////////////////////////////////////////////
/* -- AWS -- */
////////////////////////////////////////////////////
if( workflow.profile == 'awsbatch') {
// AWSBatch sanity checking
if (!params.awsqueue || !params.awsregion) exit 1, "Specify correct --awsqueue and --awsregion parameters on AWSBatch!"
// Check outdir paths to be S3 buckets if running on AWSBatch
// related: https://github.com/nextflow-io/nextflow/issues/813
if (!params.outdir.startsWith('s3:')) exit 1, "Outdir not on S3 - specify S3 Bucket to run on AWSBatch!"
// Prevent trace files to be stored on S3 since S3 does not support rolling files.
if (workflow.tracedir.startsWith('s3:')) exit 1, "Specify a local tracedir or run without trace! S3 cannot be used for tracefiles."
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- HEADER LOG INFO -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
// Header log info
log.info nfcoreHeader()
def summary = [:]
summary['Run Name'] = custom_runName ?: workflow.runName
summary['Data Type'] = params.singleEnd ? 'Single-End' : 'Paired-End'
summary['Design File'] = params.design
summary['Genome'] = params.genome ?: 'Not supplied'
summary['Fasta File'] = params.fasta
summary['GTF File'] = params.gtf
if (params.gene_bed) summary['Gene BED File'] = params.gene_bed
if (params.tss_bed) summary['TSS BED File'] = params.tss_bed
if (params.bwa_index) summary['BWA Index'] = params.bwa_index
if (params.blacklist) summary['Blacklist BED'] = params.blacklist
summary['MACS2 Genome Size'] = params.macs_gsize ?: 'Not supplied'
summary['Min Consensus Reps'] = params.min_reps_consensus
if (params.macs_gsize) summary['MACS2 Narrow Peaks'] = params.narrowPeak ? 'Yes' : 'No'
if (!params.narrowPeak) summary['MACS2 Broad Cutoff'] = params.broad_cutoff
if (params.skipTrimming){
summary['Trimming Step'] = 'Skipped'
} else {
summary['Trim R1'] = "$params.clip_r1 bp"
summary['Trim R2'] = "$params.clip_r2 bp"
summary["Trim 3' R1"] = "$params.three_prime_clip_r1 bp"
summary["Trim 3' R2"] = "$params.three_prime_clip_r2 bp"
}
if (params.seq_center) summary['Sequencing Center'] = params.seq_center
if (params.singleEnd) summary['Fragment Size'] = "$params.fragment_size bp"
summary['Fingerprint Bins'] = params.fingerprint_bins
if (params.keepDups) summary['Keep Duplicates'] = 'Yes'
if (params.keepMultiMap) summary['Keep Multi-mapped'] = 'Yes'
summary['Save Genome Index'] = params.saveGenomeIndex ? 'Yes' : 'No'
if (params.saveTrimmed) summary['Save Trimmed'] = 'Yes'
if (params.saveAlignedIntermediates) summary['Save Intermeds'] = 'Yes'
if (params.saveMACSPileup) summary['Save MACS2 Pileup'] = 'Yes'
if (params.skipDiffAnalysis) summary['Skip Diff Analysis'] = 'Yes'
if (params.skipFastQC) summary['Skip FastQC'] = 'Yes'
if (params.skipPicardMetrics) summary['Skip Picard Metrics'] = 'Yes'
if (params.skipPreseq) summary['Skip Preseq'] = 'Yes'
if (params.skipPlotProfile) summary['Skip plotProfile'] = 'Yes'
if (params.skipPlotFingerprint) summary['Skip plotFingerprint'] = 'Yes'
if (params.skipSpp) summary['Skip spp'] = 'Yes'
if (params.skipIGV) summary['Skip IGV'] = 'Yes'
if (params.skipMultiQC) summary['Skip MultiQC'] = 'Yes'
if (params.skipMultiQCStats) summary['Skip MultiQC Stats'] = 'Yes'
summary['Max Resources'] = "$params.max_memory memory, $params.max_cpus cpus, $params.max_time time per job"
if(workflow.containerEngine) summary['Container'] = "$workflow.containerEngine - $workflow.container"
summary['Output Dir'] = params.outdir
summary['Launch Dir'] = workflow.launchDir
summary['Working Dir'] = workflow.workDir
summary['Script Dir'] = workflow.projectDir
summary['User'] = workflow.userName
if (workflow.profile == 'awsbatch'){
summary['AWS Region'] = params.awsregion
summary['AWS Queue'] = params.awsqueue
}
summary['Config Profile'] = workflow.profile
if (params.config_profile_description) summary['Config Description'] = params.config_profile_description
if (params.config_profile_contact) summary['Config Contact'] = params.config_profile_contact
if (params.config_profile_url) summary['Config URL'] = params.config_profile_url
if(params.email) {
summary['E-mail Address'] = params.email
summary['MultiQC Max Size'] = params.maxMultiqcEmailFileSize
}
log.info summary.collect { k,v -> "${k.padRight(20)}: $v" }.join("\n")
log.info "\033[2m----------------------------------------------------\033[0m"
// Check the hostnames against configured profiles
checkHostname()
// Show a big warning message if we're not running MACS
if (!params.macs_gsize){
def warnstring = params.genome ? "supported for '${params.genome}'" : 'supplied'
log.warn "=================================================================\n" +
" WARNING! MACS genome size parameter not $warnstring.\n" +
" Peak calling, annotation and differential analysis will be skipped.\n" +
" Please specify value for '--macs_gsize' to run these steps.\n" +
"======================================================================="
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- PARSE DESIGN FILE -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* PREPROCESSING - REFORMAT DESIGN FILE, CHECK VALIDITY & CREATE IP vs CONTROL MAPPINGS
*/
process checkDesign {
tag "$design"
publishDir "${params.outdir}/pipeline_info", mode: 'copy'
input:
file design from ch_design
output:
file "design_reads.csv" into ch_design_reads_csv
file "design_controls.csv" into ch_design_controls_csv
script: // This script is bundled with the pipeline, in nf-core/chipseq/bin/
"""
check_design.py $design design_reads.csv design_controls.csv
"""
}
/*
* Create channels for input fastq files
*/
if (params.singleEnd) {
ch_design_reads_csv.splitCsv(header:true, sep:',')
.map { row -> [ row.sample_id, [ file(row.fastq_1, checkIfExists: true) ] ] }
.into { ch_raw_reads_fastqc;
ch_raw_reads_trimgalore }
} else {
ch_design_reads_csv.splitCsv(header:true, sep:',')
.map { row -> [ row.sample_id, [ file(row.fastq_1, checkIfExists: true), file(row.fastq_2, checkIfExists: true) ] ] }
.into { ch_raw_reads_fastqc;
ch_raw_reads_trimgalore }
}
/*
* Create a channel with [sample_id, control id, antibody, replicatesExist, multipleGroups]
*/
ch_design_controls_csv.splitCsv(header:true, sep:',')
.map { row -> [ row.sample_id, row.control_id, row.antibody, row.replicatesExist.toBoolean(), row.multipleGroups.toBoolean() ] }
.set { ch_design_controls_csv }
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- PREPARE ANNOTATION FILES -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* PREPROCESSING - Build BWA index
*/
if (!params.bwa_index){
process makeBWAindex {
tag "$fasta"
label 'process_high'
publishDir path: { params.saveGenomeIndex ? "${params.outdir}/reference_genome" : params.outdir },
saveAs: { params.saveGenomeIndex ? it : null }, mode: 'copy'
input:
file fasta from ch_fasta
output:
file "BWAIndex" into ch_bwa_index
script:
"""
bwa index -a bwtsw $fasta
mkdir BWAIndex && mv ${fasta}* BWAIndex
"""
}
}
/*
* PREPROCESSING - Generate gene BED file
*/
if (!params.gene_bed){
process makeGeneBED {
tag "$gtf"
label 'process_low'
publishDir "${params.outdir}/reference_genome", mode: 'copy'
input:
file gtf from ch_gtf
output:
file "*.bed" into ch_gene_bed
script: // This script is bundled with the pipeline, in nf-core/chipseq/bin/
"""
gtf2bed $gtf > ${gtf.baseName}.bed
"""
}
}
/*
* PREPROCESSING - Generate TSS BED file
*/
if (!params.tss_bed){
process makeTSSBED {
tag "$bed"
publishDir "${params.outdir}/reference_genome", mode: 'copy'
input:
file bed from ch_gene_bed
output:
file "*.bed" into ch_tss_bed
script:
"""
cat $bed | awk -v FS='\t' -v OFS='\t' '{ if(\$6=="+") \$3=\$2+1; else \$2=\$3-1; print \$1, \$2, \$3, \$4, \$5, \$6;}' > ${bed.baseName}.tss.bed
"""
}
}
/*
* PREPROCESSING - Prepare genome intervals for filtering
*/
process makeGenomeFilter {
tag "$fasta"
publishDir "${params.outdir}/reference_genome", mode: 'copy'
input:
file fasta from ch_fasta
output:
file "$fasta" into ch_genome_fasta // FASTA FILE FOR IGV
file "*.fai" into ch_genome_fai // FAI INDEX FOR REFERENCE GENOME
file "*.bed" into ch_genome_filter_regions // BED FILE WITHOUT BLACKLIST REGIONS
file "*.sizes" into ch_genome_sizes_bigwig // CHROMOSOME SIZES FILE FOR BEDTOOLS
script:
blacklist_filter = params.blacklist ? "sortBed -i ${params.blacklist} -g ${fasta}.sizes | complementBed -i stdin -g ${fasta}.sizes" : "awk '{print \$1, '0' , \$2}' OFS='\t' ${fasta}.sizes"
"""
samtools faidx $fasta
cut -f 1,2 ${fasta}.fai > ${fasta}.sizes
$blacklist_filter > ${fasta}.include_regions.bed
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- FASTQ QC -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 1 - FastQC
*/
process fastqc {
tag "$name"
label 'process_medium'
publishDir "${params.outdir}/fastqc", mode: 'copy',
saveAs: {filename -> filename.endsWith(".zip") ? "zips/$filename" : "$filename"}
when:
!params.skipFastQC
input:
set val(name), file(reads) from ch_raw_reads_fastqc
output:
file "*.{zip,html}" into ch_fastqc_reports_mqc
script:
// Added soft-links to original fastqs for consistent naming in MultiQC
if (params.singleEnd) {
"""
[ ! -f ${name}.fastq.gz ] && ln -s $reads ${name}.fastq.gz
fastqc -q ${name}.fastq.gz
"""
} else {
"""
[ ! -f ${name}_1.fastq.gz ] && ln -s ${reads[0]} ${name}_1.fastq.gz
[ ! -f ${name}_2.fastq.gz ] && ln -s ${reads[1]} ${name}_2.fastq.gz
fastqc -q ${name}_1.fastq.gz
fastqc -q ${name}_2.fastq.gz
"""
}
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- ADAPTER TRIMMING -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 2 - Trim Galore!
*/
if (params.skipTrimming){
ch_trimmed_reads = ch_raw_reads_trimgalore
ch_trimgalore_results_mqc = []
ch_trimgalore_fastqc_reports_mqc = []
} else {
process trimGalore {
tag "$name"
label 'process_long'
publishDir "${params.outdir}/trim_galore", mode: 'copy',
saveAs: {filename ->
if (filename.endsWith(".html")) "fastqc/$filename"
else if (filename.endsWith(".zip")) "fastqc/zip/$filename"
else if (filename.endsWith("trimming_report.txt")) "logs/$filename"
else params.saveTrimmed ? filename : null
}
input:
set val(name), file(reads) from ch_raw_reads_trimgalore
output:
set val(name), file("*.fq.gz") into ch_trimmed_reads
file "*.txt" into ch_trimgalore_results_mqc
file "*.{zip,html}" into ch_trimgalore_fastqc_reports_mqc
script:
// Added soft-links to original fastqs for consistent naming in MultiQC
c_r1 = params.clip_r1 > 0 ? "--clip_r1 ${params.clip_r1}" : ''
c_r2 = params.clip_r2 > 0 ? "--clip_r2 ${params.clip_r2}" : ''
tpc_r1 = params.three_prime_clip_r1 > 0 ? "--three_prime_clip_r1 ${params.three_prime_clip_r1}" : ''
tpc_r2 = params.three_prime_clip_r2 > 0 ? "--three_prime_clip_r2 ${params.three_prime_clip_r2}" : ''
if (params.singleEnd) {
"""
[ ! -f ${name}.fastq.gz ] && ln -s $reads ${name}.fastq.gz
trim_galore --fastqc --gzip $c_r1 $tpc_r1 ${name}.fastq.gz
"""
} else {
"""
[ ! -f ${name}_1.fastq.gz ] && ln -s ${reads[0]} ${name}_1.fastq.gz
[ ! -f ${name}_2.fastq.gz ] && ln -s ${reads[1]} ${name}_2.fastq.gz
trim_galore --paired --fastqc --gzip $c_r1 $c_r2 $tpc_r1 $tpc_r2 ${name}_1.fastq.gz ${name}_2.fastq.gz
"""
}
}
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- ALIGN -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 3.1 - Align read 1 with bwa
*/
process bwaMEM {
tag "$name"
label 'process_high'
input:
set val(name), file(reads) from ch_trimmed_reads
file index from ch_bwa_index.collect()
output:
set val(name), file("*.bam") into ch_bwa_bam
script:
prefix="${name}.Lb"
if (!params.seq_center) {
rg="\'@RG\\tID:${name}\\tSM:${name.split('_')[0..-2].join('_')}\\tPL:ILLUMINA\\tLB:${name}\\tPU:1\'"
} else {
rg="\'@RG\\tID:${name}\\tSM:${name.split('_')[0..-2].join('_')}\\tPL:ILLUMINA\\tLB:${name}\\tPU:1\\tCN:${params.seq_center}\'"
}
"""
bwa mem \\
-t $task.cpus \\
-M \\
-R $rg \\
${index}/${bwa_base} \\
$reads \\
| samtools view -@ $task.cpus -b -h -F 0x0100 -O BAM -o ${prefix}.bam -
"""
}
/*
* STEP 3.2 - Convert .bam to coordinate sorted .bam
*/
process sortBAM {
tag "$name"
label 'process_medium'
if (params.saveAlignedIntermediates) {
publishDir path: "${params.outdir}/bwa/library", mode: 'copy',
saveAs: { filename ->
if (filename.endsWith(".flagstat")) "samtools_stats/$filename"
else if (filename.endsWith(".idxstats")) "samtools_stats/$filename"
else if (filename.endsWith(".stats")) "samtools_stats/$filename"
else filename }
}
input:
set val(name), file(bam) from ch_bwa_bam
output:
set val(name), file("*.sorted.{bam,bam.bai}") into ch_sort_bam_merge
file "*.{flagstat,idxstats,stats}" into ch_sort_bam_flagstat_mqc
script:
prefix="${name}.Lb"
"""
samtools sort -@ $task.cpus -o ${prefix}.sorted.bam -T $name $bam
samtools index ${prefix}.sorted.bam
samtools flagstat ${prefix}.sorted.bam > ${prefix}.sorted.bam.flagstat
samtools idxstats ${prefix}.sorted.bam > ${prefix}.sorted.bam.idxstats
samtools stats ${prefix}.sorted.bam > ${prefix}.sorted.bam.stats
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- MERGE LIBRARY BAM -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 4.1 Merge BAM files for all libraries from same sample
*/
ch_sort_bam_merge.map { it -> [ it[0].split('_')[0..-2].join('_'), it[1] ] }
.groupTuple(by: [0])
.map { it -> [ it[0], it[1].flatten() ] }
.set { ch_sort_bam_merge }
process mergeBAM {
tag "$name"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary", mode: 'copy',
saveAs: { filename ->
if (filename.endsWith(".flagstat")) "samtools_stats/$filename"
else if (filename.endsWith(".idxstats")) "samtools_stats/$filename"
else if (filename.endsWith(".stats")) "samtools_stats/$filename"
else if (filename.endsWith(".metrics.txt")) "picard_metrics/$filename"
else params.saveAlignedIntermediates ? filename : null
}
input:
set val(name), file(bams) from ch_sort_bam_merge
output:
set val(name), file("*${prefix}.sorted.{bam,bam.bai}") into ch_merge_bam_filter,
ch_merge_bam_preseq
file "*.{flagstat,idxstats,stats}" into ch_merge_bam_stats_mqc
file "*.txt" into ch_merge_bam_metrics_mqc
script:
prefix="${name}.mLb.mkD"
bam_files = bams.findAll { it.toString().endsWith('.bam') }.sort()
if (!task.memory){
log.info "[Picard MarkDuplicates] Available memory not known - defaulting to 3GB. Specify process memory requirements to change this."
avail_mem = 3
} else {
avail_mem = task.memory.toGiga()
}
if (bam_files.size() > 1) {
"""
picard -Xmx${avail_mem}g MergeSamFiles \\
${'INPUT='+bam_files.join(' INPUT=')} \\
OUTPUT=${name}.sorted.bam \\
SORT_ORDER=coordinate \\
VALIDATION_STRINGENCY=LENIENT \\
TMP_DIR=tmp
samtools index ${name}.sorted.bam
picard -Xmx${avail_mem}g MarkDuplicates \\
INPUT=${name}.sorted.bam \\
OUTPUT=${prefix}.sorted.bam \\
ASSUME_SORTED=true \\
REMOVE_DUPLICATES=false \\
METRICS_FILE=${prefix}.MarkDuplicates.metrics.txt \\
VALIDATION_STRINGENCY=LENIENT \\
TMP_DIR=tmp
samtools index ${prefix}.sorted.bam
samtools idxstats ${prefix}.sorted.bam > ${prefix}.sorted.bam.idxstats
samtools flagstat ${prefix}.sorted.bam > ${prefix}.sorted.bam.flagstat
samtools stats ${prefix}.sorted.bam > ${prefix}.sorted.bam.stats
"""
} else {
"""
picard -Xmx${avail_mem}g MarkDuplicates \\
INPUT=${bam_files[0]} \\
OUTPUT=${prefix}.sorted.bam \\
ASSUME_SORTED=true \\
REMOVE_DUPLICATES=false \\
METRICS_FILE=${prefix}.MarkDuplicates.metrics.txt \\
VALIDATION_STRINGENCY=LENIENT \\
TMP_DIR=tmp
samtools index ${prefix}.sorted.bam
samtools idxstats ${prefix}.sorted.bam > ${prefix}.sorted.bam.idxstats
samtools flagstat ${prefix}.sorted.bam > ${prefix}.sorted.bam.flagstat
samtools stats ${prefix}.sorted.bam > ${prefix}.sorted.bam.stats
"""
}
}
/*
* STEP 4.2 Filter BAM file at merged library-level
*/
process filterBAM {
tag "$name"
label 'process_medium'
publishDir path: "${params.outdir}/bwa/mergedLibrary", mode: 'copy',
saveAs: { filename ->
if (params.singleEnd || params.saveAlignedIntermediates) {
if (filename.endsWith(".flagstat")) "samtools_stats/$filename"
else if (filename.endsWith(".idxstats")) "samtools_stats/$filename"
else if (filename.endsWith(".stats")) "samtools_stats/$filename"
else if (filename.endsWith(".sorted.bam")) filename
else if (filename.endsWith(".sorted.bam.bai")) filename
else null }
}
input:
set val(name), file(bam) from ch_merge_bam_filter
file bed from ch_genome_filter_regions.collect()
file bamtools_filter_config from ch_bamtools_filter_config
output:
set val(name), file("*.{bam,bam.bai}") into ch_filter_bam
set val(name), file("*.flagstat") into ch_filter_bam_flagstat
file "*.{idxstats,stats}" into ch_filter_bam_stats_mqc
script:
prefix = params.singleEnd ? "${name}.mLb.clN" : "${name}.mLb.flT"
filter_params = params.singleEnd ? "-F 0x004" : "-F 0x004 -F 0x0008 -f 0x001"
dup_params = params.keepDups ? "" : "-F 0x0400"
multimap_params = params.keepMultiMap ? "" : "-q 1"
blacklist_params = params.blacklist ? "-L $bed" : ""
name_sort_bam = params.singleEnd ? "" : "samtools sort -n -@ $task.cpus -o ${prefix}.bam -T $prefix ${prefix}.sorted.bam"
"""
samtools view \\
$filter_params \\
$dup_params \\
$multimap_params \\
$blacklist_params \\
-b ${bam[0]} \\
| bamtools filter \\
-out ${prefix}.sorted.bam \\
-script $bamtools_filter_config
samtools index ${prefix}.sorted.bam
samtools flagstat ${prefix}.sorted.bam > ${prefix}.sorted.bam.flagstat
samtools idxstats ${prefix}.sorted.bam > ${prefix}.sorted.bam.idxstats
samtools stats ${prefix}.sorted.bam > ${prefix}.sorted.bam.stats
$name_sort_bam
"""
}
/*
* STEP 4.3 Remove orphan reads from paired-end BAM file
*/
if (params.singleEnd){
ch_filter_bam.into { ch_rm_orphan_bam_metrics;
ch_rm_orphan_bam_bigwig;
ch_rm_orphan_bam_macs_1;
ch_rm_orphan_bam_macs_2;
ch_rm_orphan_bam_phantompeakqualtools;
ch_rm_orphan_name_bam_counts }
ch_filter_bam_flagstat.into { ch_rm_orphan_flagstat_bigwig;
ch_rm_orphan_flagstat_macs;
ch_rm_orphan_flagstat_mqc }
ch_filter_bam_stats_mqc.set { ch_rm_orphan_stats_mqc }
} else {
process rmOrphanReads {
tag "$name"
label 'process_medium'
publishDir path: "${params.outdir}/bwa/mergedLibrary", mode: 'copy',
saveAs: { filename ->
if (filename.endsWith(".flagstat")) "samtools_stats/$filename"
else if (filename.endsWith(".idxstats")) "samtools_stats/$filename"
else if (filename.endsWith(".stats")) "samtools_stats/$filename"
else if (filename.endsWith(".sorted.bam")) filename
else if (filename.endsWith(".sorted.bam.bai")) filename
else null
}
input:
set val(name), file(bam) from ch_filter_bam
output:
set val(name), file("*.sorted.{bam,bam.bai}") into ch_rm_orphan_bam_metrics,
ch_rm_orphan_bam_bigwig,
ch_rm_orphan_bam_macs_1,
ch_rm_orphan_bam_macs_2,
ch_rm_orphan_bam_phantompeakqualtools
set val(name), file("${prefix}.bam") into ch_rm_orphan_name_bam_counts
set val(name), file("*.flagstat") into ch_rm_orphan_flagstat_bigwig,
ch_rm_orphan_flagstat_macs,
ch_rm_orphan_flagstat_mqc
file "*.{idxstats,stats}" into ch_rm_orphan_stats_mqc
script: // This script is bundled with the pipeline, in nf-core/chipseq/bin/
prefix="${name}.mLb.clN"
"""
bampe_rm_orphan.py ${bam[0]} ${prefix}.bam --only_fr_pairs
samtools sort -@ $task.cpus -o ${prefix}.sorted.bam -T $prefix ${prefix}.bam
samtools index ${prefix}.sorted.bam
samtools flagstat ${prefix}.sorted.bam > ${prefix}.sorted.bam.flagstat
samtools idxstats ${prefix}.sorted.bam > ${prefix}.sorted.bam.idxstats
samtools stats ${prefix}.sorted.bam > ${prefix}.sorted.bam.stats
"""
}
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- MERGE LIBRARY BAM POST-ANALYSIS -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 5.1 preseq analysis after merging libraries and before filtering
*/
process preseq {
tag "$name"
label 'process_low'
publishDir "${params.outdir}/bwa/mergedLibrary/preseq", mode: 'copy'
when:
!params.skipPreseq
input:
set val(name), file(bam) from ch_merge_bam_preseq
output:
file "*.ccurve.txt" into ch_preseq_results
script:
prefix="${name}.mLb.clN"
"""
preseq lc_extrap -v -output ${prefix}.ccurve.txt -bam ${bam[0]}
"""
}
/*
* STEP 5.2 Picard CollectMultipleMetrics after merging libraries and filtering
*/
process collectMultipleMetrics {
tag "$name"
label 'process_medium'
publishDir path: "${params.outdir}/bwa/mergedLibrary", mode: 'copy',
saveAs: { filename ->
if (filename.endsWith("_metrics")) "picard_metrics/$filename"
else if (filename.endsWith(".pdf")) "picard_metrics/pdf/$filename"
else null
}
when:
!params.skipPicardMetrics
input:
set val(name), file(bam) from ch_rm_orphan_bam_metrics
file fasta from ch_fasta
output:
file "*_metrics" into ch_collectmetrics_mqc
file "*.pdf" into ch_collectmetrics_pdf
script:
prefix="${name}.mLb.clN"
if (!task.memory){
log.info "[Picard CollectMultipleMetrics] Available memory not known - defaulting to 3GB. Specify process memory requirements to change this."
avail_mem = 3
} else {
avail_mem = task.memory.toGiga()
}
"""
picard -Xmx${avail_mem}g CollectMultipleMetrics \\
INPUT=${bam[0]} \\
OUTPUT=${prefix}.CollectMultipleMetrics \\
REFERENCE_SEQUENCE=$fasta \\
VALIDATION_STRINGENCY=LENIENT \\
TMP_DIR=tmp
"""
}
/*
* STEP 5.3 Read depth normalised bigWig
*/
process bigWig {
tag "$name"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary/bigwig", mode: 'copy',
saveAs: {filename ->
if (filename.endsWith(".txt")) "scale/$filename"
else if (filename.endsWith(".bigWig")) "$filename"
else null
}
input:
set val(name), file(bam), file(flagstat) from ch_rm_orphan_bam_bigwig.join(ch_rm_orphan_flagstat_bigwig, by: [0])
file sizes from ch_genome_sizes_bigwig.collect()
output:
set val(name), file("*.bigWig") into ch_bigwig_plotprofile
file "*scale_factor.txt" into ch_bigwig_scale
file "*igv.txt" into ch_bigwig_igv
script:
prefix="${name}.mLb.clN"
pe_fragment = params.singleEnd ? "" : "-pc"
extend = (params.singleEnd && params.fragment_size > 0) ? "-fs ${params.fragment_size}" : ''
"""
SCALE_FACTOR=\$(grep 'mapped (' $flagstat | awk '{print 1000000/\$1}')
echo \$SCALE_FACTOR > ${prefix}.scale_factor.txt
genomeCoverageBed -ibam ${bam[0]} -bg -scale \$SCALE_FACTOR $pe_fragment $extend | sort -k1,1 -k2,2n > ${prefix}.bedGraph
bedGraphToBigWig ${prefix}.bedGraph $sizes ${prefix}.bigWig
find * -type f -name "*.bigWig" -exec echo -e "bwa/mergedLibrary/bigwig/"{}"\\t0,0,178" \\; > ${prefix}.bigWig.igv.txt
"""
}
/*
* STEP 5.4 generate gene body coverage plot with deepTools
*/
process plotProfile {
tag "$name"
label 'process_high'
publishDir "${params.outdir}/bwa/mergedLibrary/deepTools/plotProfile", mode: 'copy'
when:
!params.skipPlotProfile
input:
set val(name), file(bigwig) from ch_bigwig_plotprofile
file bed from ch_gene_bed
output:
file '*.{gz,pdf}' into ch_plotprofile_results
file '*.plotProfile.tab' into ch_plotprofile_mqc
script:
"""
computeMatrix scale-regions \\
--regionsFileName $bed \\
--scoreFileName $bigwig \\
--outFileName ${name}.computeMatrix.mat.gz \\
--outFileNameMatrix ${name}.computeMatrix.vals.mat.gz \\
--regionBodyLength 1000 \\
--beforeRegionStartLength 3000 \\
--afterRegionStartLength 3000 \\
--skipZeros \\
--smartLabels \\
-p $task.cpus
plotProfile --matrixFile ${name}.computeMatrix.mat.gz \\
--outFileName ${name}.plotProfile.pdf \\
--outFileNameData ${name}.plotProfile.tab
"""
}
/*
* STEP 5.5 Phantompeakqualtools
*/
process phantomPeakQualTools {
tag "$name"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary/phantompeakqualtools", mode: 'copy'
when:
!params.skipSpp
input:
set val(name), file(bam) from ch_rm_orphan_bam_phantompeakqualtools
file spp_correlation_header from ch_spp_correlation_header
file spp_nsc_header from ch_spp_nsc_header
file spp_rsc_header from ch_spp_rsc_header
output:
file '*.pdf' into ch_spp_plot
file '*.spp.out' into ch_spp_out,
ch_spp_out_mqc
file '*_mqc.tsv' into ch_spp_csv_mqc
script:
"""
RUN_SPP=`which run_spp.R`
Rscript -e "library(caTools); source(\\"\$RUN_SPP\\")" -c="${bam[0]}" -savp="${name}.spp.pdf" -savd="${name}.spp.Rdata" -out="${name}.spp.out" -p=$task.cpus
cp $spp_correlation_header ${name}_spp_correlation_mqc.tsv
Rscript -e "load('${name}.spp.Rdata'); write.table(crosscorr\\\$cross.correlation, file=\\"${name}_spp_correlation_mqc.tsv\\", sep=",", quote=FALSE, row.names=FALSE, col.names=FALSE,append=TRUE)"
awk -v OFS='\t' '{print "${name}", \$9}' ${name}.spp.out | cat $spp_nsc_header - > ${name}_spp_nsc_mqc.tsv
awk -v OFS='\t' '{print "${name}", \$10}' ${name}.spp.out | cat $spp_rsc_header - > ${name}_spp_rsc_mqc.tsv
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- MERGE LIBRARY PEAK ANALYSIS -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
// Create channel linking IP bams with control bams
ch_rm_orphan_bam_macs_1.combine(ch_rm_orphan_bam_macs_2)
.set { ch_rm_orphan_bam_macs_1 }
ch_design_controls_csv.combine(ch_rm_orphan_bam_macs_1)
.filter { it[0] == it[5] && it[1] == it[7] }
.join(ch_rm_orphan_flagstat_macs)
.map { it -> it[2..-1] }
.into { ch_group_bam_macs;
ch_group_bam_plotfingerprint;
ch_group_bam_deseq }
/*
* STEP 6.1 deepTools plotFingerprint
*/
process plotFingerprint {
tag "${ip} vs ${control}"
label 'process_high'
publishDir "${params.outdir}/bwa/mergedLibrary/deepTools/plotFingerprint", mode: 'copy'
when:
!params.skipPlotFingerprint
input:
set val(antibody), val(replicatesExist), val(multipleGroups), val(ip), file(ipbam), val(control), file(controlbam), file(ipflagstat) from ch_group_bam_plotfingerprint
output:
file '*.{txt,pdf}' into ch_plotfingerprint_results
file '*.raw.txt' into ch_plotfingerprint_mqc
script:
extend = (params.singleEnd && params.fragment_size > 0) ? "--extendReads ${params.fragment_size}" : ''
"""
plotFingerprint \\
--bamfiles ${ipbam[0]} ${controlbam[0]} \\
--plotFile ${ip}.plotFingerprint.pdf \\
$extend \\
--labels $ip $control \\
--outRawCounts ${ip}.plotFingerprint.raw.txt \\
--outQualityMetrics ${ip}.plotFingerprint.qcmetrics.txt \\
--skipZeros \\
--JSDsample ${controlbam[0]} \\
--numberOfProcessors ${task.cpus} \\
--numberOfSamples ${params.fingerprint_bins}
"""
}
/*
* STEP 6.2 Call peaks with MACS2 and calculate FRiP score
*/
process macsCallPeak {
tag "${ip} vs ${control}"
label 'process_long'
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}", mode: 'copy',
saveAs: {filename ->
if (filename.endsWith(".tsv")) "qc/$filename"
else if (filename.endsWith(".igv.txt")) null
else filename
}
when:
params.macs_gsize
input:
set val(antibody), val(replicatesExist), val(multipleGroups), val(ip), file(ipbam), val(control), file(controlbam), file(ipflagstat) from ch_group_bam_macs
file peak_count_header from ch_peak_count_header
file frip_score_header from ch_frip_score_header
output:
set val(ip), file("*.{bed,xls,gappedPeak,bdg}") into ch_macs_output
set val(antibody), val(replicatesExist), val(multipleGroups), val(ip), val(control), file("*.$peaktype") into ch_macs_homer, ch_macs_qc, ch_macs_consensus
file "*igv.txt" into ch_macs_igv
file "*_mqc.tsv" into ch_macs_mqc
script:
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
broad = params.narrowPeak ? '' : "--broad --broad-cutoff ${params.broad_cutoff}"
format = params.singleEnd ? "BAM" : "BAMPE"
pileup = params.saveMACSPileup ? "-B --SPMR" : ""
"""
macs2 callpeak \\
-t ${ipbam[0]} \\
-c ${controlbam[0]} \\
$broad \\
-f $format \\
-g ${params.macs_gsize} \\
-n $ip \\
$pileup \\
--keep-dup all \\
--nomodel
cat ${ip}_peaks.${peaktype} | wc -l | awk -v OFS='\t' '{ print "${ip}", \$1 }' | cat $peak_count_header - > ${ip}_peaks.count_mqc.tsv
READS_IN_PEAKS=\$(intersectBed -a ${ipbam[0]} -b ${ip}_peaks.${peaktype} -bed -c -f 0.20 | awk -F '\t' '{sum += \$NF} END {print sum}')
grep 'mapped (' $ipflagstat | awk -v a="\$READS_IN_PEAKS" -v OFS='\t' '{print "${ip}", a/\$1}' | cat $frip_score_header - > ${ip}_peaks.FRiP_mqc.tsv
find * -type f -name "*.${peaktype}" -exec echo -e "bwa/mergedLibrary/macs/${peaktype}/"{}"\\t0,0,178" \\; > ${ip}_peaks.${peaktype}.igv.txt
"""
}
/*
* STEP 6.3 Annotate peaks with HOMER
*/
process annotatePeaks {
tag "${ip} vs ${control}"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}", mode: 'copy'
when:
params.macs_gsize
input:
set val(antibody), val(replicatesExist), val(multipleGroups), val(ip), val(control), file(peak) from ch_macs_homer
file fasta from ch_fasta
file gtf from ch_gtf
output:
file "*.txt" into ch_macs_annotate
script:
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
"""
annotatePeaks.pl $peak \\
$fasta \\
-gid \\
-gtf $gtf \\
> ${ip}_peaks.annotatePeaks.txt
"""
}
/*
* STEP 6.4 Aggregated QC plots for peaks, FRiP and peak-to-gene annotation
*/
process peakQC {
label "process_medium"
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}/qc", mode: 'copy'
when:
params.macs_gsize
input:
file peaks from ch_macs_qc.collect{ it[-1] }
file annos from ch_macs_annotate.collect()
file peak_annotation_header from ch_peak_annotation_header
output:
file "*.{txt,pdf}" into ch_macs_qc_output
file "*.tsv" into ch_macs_qc_mqc
script: // This script is bundled with the pipeline, in nf-core/chipseq/bin/
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
"""
plot_macs_qc.r -i ${peaks.join(',')} \\
-s ${peaks.join(',').replaceAll("_peaks.${peaktype}","")} \\
-o ./ \\
-p macs_peak
plot_homer_annotatepeaks.r -i ${annos.join(',')} \\
-s ${annos.join(',').replaceAll("_peaks.annotatePeaks.txt","")} \\
-o ./ \\
-p macs_annotatePeaks
cat $peak_annotation_header macs_annotatePeaks.summary.txt > macs_annotatePeaks.summary_mqc.tsv
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- CONSENSUS PEAKS ANALYSIS -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
// group by ip from this point and carry forward boolean variables
ch_macs_consensus.map { it -> [ it[0], it[1], it[2], it[-1] ] }
.groupTuple()
.map { it -> [ it[0], it[1][0], it[2][0], it[3].sort() ] }
.set { ch_macs_consensus }
/*
* STEP 7.1 Consensus peaks across samples, create boolean filtering file, .saf file for featureCounts and UpSetR plot for intersection
*/
process createConsensusPeakSet {
tag "${antibody}"
label 'process_long'
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}/consensus/${antibody}", mode: 'copy',
saveAs: {filename ->
if (filename.endsWith(".igv.txt")) null
else filename
}
when:
params.macs_gsize && (replicatesExist || multipleGroups)
input:
set val(antibody), val(replicatesExist), val(multipleGroups), file(peaks) from ch_macs_consensus
output:
set val(antibody), val(replicatesExist), val(multipleGroups), file("*.bed") into ch_macs_consensus_bed
set val(antibody), file("*.saf") into ch_macs_consensus_saf
file "*.boolean.txt" into ch_macs_consensus_bool
file "*.intersect.{txt,plot.pdf}" into ch_macs_consensus_intersect
file "*igv.txt" into ch_macs_consensus_igv
script: // scripts are bundled with the pipeline, in nf-core/chipseq/bin/
prefix="${antibody}.consensus_peaks"
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
mergecols = params.narrowPeak ? (2..10).join(',') : (2..9).join(',')
collapsecols = params.narrowPeak ? (["collapse"]*9).join(',') : (["collapse"]*8).join(',')
expandparam = params.narrowPeak ? "--is_narrow_peak" : ""
"""
sort -k1,1 -k2,2n ${peaks.collect{it.toString()}.sort().join(' ')} \\
| mergeBed -c $mergecols -o $collapsecols > ${prefix}.txt
macs2_merged_expand.py ${prefix}.txt \\
${peaks.collect{it.toString()}.sort().join(',').replaceAll("_peaks.${peaktype}","")} \\
${prefix}.boolean.txt \\
--min_replicates $params.min_reps_consensus \\
$expandparam
awk -v FS='\t' -v OFS='\t' 'FNR > 1 { print \$1, \$2, \$3, \$4, "0", "+" }' ${prefix}.boolean.txt > ${prefix}.bed
echo -e "GeneID\tChr\tStart\tEnd\tStrand" > ${prefix}.saf
awk -v FS='\t' -v OFS='\t' 'FNR > 1 { print \$4, \$1, \$2, \$3, "+" }' ${prefix}.boolean.txt >> ${prefix}.saf
plot_peak_intersect.r -i ${prefix}.boolean.intersect.txt -o ${prefix}.boolean.intersect.plot.pdf
find * -type f -name "${prefix}.bed" -exec echo -e "bwa/mergedLibrary/macs/${peaktype}/consensus/${antibody}/"{}"\\t0,0,0" \\; > ${prefix}.bed.igv.txt
"""
}
/*
* STEP 7.2 Annotate consensus peaks with HOMER, and add annotation to boolean output file
*/
process annotateConsensusPeakSet {
tag "${antibody}"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}/consensus/${antibody}", mode: 'copy'
when:
params.macs_gsize && (replicatesExist || multipleGroups)
input:
set val(antibody), val(replicatesExist), val(multipleGroups), file(bed) from ch_macs_consensus_bed
file bool from ch_macs_consensus_bool
file fasta from ch_fasta
file gtf from ch_gtf
output:
file "*.annotatePeaks.txt" into ch_macs_consensus_annotate
script:
prefix="${antibody}.consensus_peaks"
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
"""
annotatePeaks.pl $bed \\
$fasta \\
-gid \\
-gtf $gtf \\
> ${prefix}.annotatePeaks.txt
cut -f2- ${prefix}.annotatePeaks.txt | awk 'NR==1; NR > 1 {print \$0 | "sort -k1,1 -k2,2n"}' | cut -f6- > tmp.txt
paste $bool tmp.txt > ${prefix}.boolean.annotatePeaks.txt
"""
}
// get bam and saf files for each ip
ch_group_bam_deseq.map { it -> [ it[3], [ it[0], it[1], it[2] ] ] }
.join(ch_rm_orphan_name_bam_counts)
.map { it -> [ it[1][0], it[1][1], it[1][2], it[2] ] }
.groupTuple()
.map { it -> [ it[0], it[1][0], it[2][0], it[3].flatten().sort() ] }
.join(ch_macs_consensus_saf)
.set { ch_group_bam_deseq }
/*
* STEP 7.3 Count reads in consensus peaks with featureCounts and perform differential analysis with DESeq2
*/
process deseqConsensusPeakSet {
tag "${antibody}"
label 'process_medium'
publishDir "${params.outdir}/bwa/mergedLibrary/macs/${peaktype}/consensus/${antibody}/deseq2", mode: 'copy',
saveAs: {filename ->
if (filename.endsWith(".igv.txt")) null
else filename
}
when:
params.macs_gsize && !params.skipDiffAnalysis && replicatesExist && multipleGroups
input:
set val(antibody), val(replicatesExist), val(multipleGroups), file(bams) ,file(saf) from ch_group_bam_deseq
file deseq2_pca_header from ch_deseq2_pca_header
file deseq2_clustering_header from ch_deseq2_clustering_header
output:
file "*featureCounts.txt" into ch_macs_consensus_counts
file "*featureCounts.txt.summary" into ch_macs_consensus_counts_mqc
file "*.{RData,results.txt,pdf,log}" into ch_macs_consensus_deseq_results
file "sizeFactors" into ch_macs_consensus_deseq_factors
file "*vs*/*.{pdf,txt}" into ch_macs_consensus_deseq_comp_results
file "*vs*/*.bed" into ch_macs_consensus_deseq_comp_bed
file "*igv.txt" into ch_macs_consensus_deseq_comp_igv
file "*.tsv" into ch_macs_consensus_deseq_mqc
script:
prefix="${antibody}.consensus_peaks"
peaktype = params.narrowPeak ? "narrowPeak" : "broadPeak"
bam_files = bams.findAll { it.toString().endsWith('.bam') }.sort()
bam_ext = params.singleEnd ? ".mLb.clN.sorted.bam" : ".mLb.clN.bam"
pe_params = params.singleEnd ? '' : "-p --donotsort"
"""
featureCounts -F SAF \\
-O \\
--fracOverlap 0.2 \\
-T $task.cpus \\
$pe_params \\
-a $saf \\
-o ${prefix}.featureCounts.txt \\
${bam_files.join(' ')}
featurecounts_deseq2.r -i ${prefix}.featureCounts.txt -b '$bam_ext' -o ./ -p $prefix -s .mLb
sed 's/deseq2_pca/deseq2_pca_${task.index}/g' <$deseq2_pca_header >tmp.txt
sed -i -e 's/DESeq2:/${antibody} DESeq2:/g' tmp.txt
cat tmp.txt ${prefix}.pca.vals.txt > ${prefix}.pca.vals_mqc.tsv
sed 's/deseq2_clustering/deseq2_clustering_${task.index}/g' <$deseq2_clustering_header >tmp.txt
sed -i -e 's/DESeq2:/${antibody} DESeq2:/g' tmp.txt
cat tmp.txt ${prefix}.sample.dists.txt > ${prefix}.sample.dists_mqc.tsv
find * -type f -name "*.FDR0.05.results.bed" -exec echo -e "bwa/mergedLibrary/macs/${peaktype}/consensus/${antibody}/deseq2/"{}"\\t255,0,0" \\; > ${prefix}.igv.txt
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- IGV -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 8 - Create IGV session file
*/
process igv {
publishDir "${params.outdir}/igv", mode: 'copy'
when:
!params.skipIGV
input:
file fasta from ch_fasta
file bigwigs from ch_bigwig_igv.collect().ifEmpty([])
file peaks from ch_macs_igv.collect().ifEmpty([])
file consensus_peaks from ch_macs_consensus_igv.collect().ifEmpty([])
file differential_peaks from ch_macs_consensus_deseq_comp_igv.collect().ifEmpty([])
output:
file "*.{txt,xml}" into ch_igv_session
script: // scripts are bundled with the pipeline, in nf-core/chipseq/bin/
"""
cat *.txt > igv_files.txt
igv_files_to_session.py igv_session.xml igv_files.txt ../reference_genome/${fasta.getName()} --path_prefix '../'
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- MULTIQC -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* Parse software version numbers
*/
process get_software_versions {
publishDir "${params.outdir}/pipeline_info", mode: 'copy',
saveAs: {filename ->
if (filename.indexOf(".csv") > 0) filename
else null
}
output:
file 'software_versions_mqc.yaml' into ch_software_versions_mqc
file "software_versions.csv"
script:
"""
echo $workflow.manifest.version > v_pipeline.txt
echo $workflow.nextflow.version > v_nextflow.txt
fastqc --version > v_fastqc.txt
trim_galore --version > v_trim_galore.txt
echo \$(bwa 2>&1) > v_bwa.txt
samtools --version > v_samtools.txt
bedtools --version > v_bedtools.txt
echo \$(bamtools --version 2>&1) > v_bamtools.txt
echo \$(plotFingerprint --version 2>&1) > v_deeptools.txt || true
picard MarkDuplicates --version &> v_picard.txt || true
echo \$(R --version 2>&1) > v_R.txt
python -c "import pysam; print(pysam.__version__)" > v_pysam.txt
echo \$(macs2 --version 2>&1) > v_macs2.txt
touch v_homer.txt
echo \$(featureCounts -v 2>&1) > v_featurecounts.txt
preseq &> v_preseq.txt
multiqc --version > v_multiqc.txt
scrape_software_versions.py &> software_versions_mqc.yaml
"""
}
def create_workflow_summary(summary) {
def yaml_file = workDir.resolve('workflow_summary_mqc.yaml')
yaml_file.text = """
id: 'nf-core-chipseq-summary'
description: " - this information is collected when the pipeline is started."
section_name: 'nf-core/chipseq Workflow Summary'
section_href: 'https://github.com/nf-core/chipseq'
plot_type: 'html'
data: |
<dl class=\"dl-horizontal\">
${summary.collect { k,v -> " <dt>$k</dt><dd><samp>${v ?: '<span style=\"color:#999999;\">N/A</a>'}</samp></dd>" }.join("\n")}
</dl>
""".stripIndent()
return yaml_file
}
/*
* STEP 9 - MultiQC
*/
process multiqc {
publishDir "${params.outdir}/multiqc", mode: 'copy'
when:
!params.skipMultiQC
input:
file multiqc_config from ch_multiqc_config
file ('fastqc/*') from ch_fastqc_reports_mqc.collect()
file ('trimgalore/*') from ch_trimgalore_results_mqc.collect()
file ('trimgalore/fastqc/*') from ch_trimgalore_fastqc_reports_mqc.collect()
file ('alignment/library/*') from ch_sort_bam_flagstat_mqc.collect()
file ('alignment/mergedLibrary/*') from ch_merge_bam_stats_mqc.collect()
file ('alignment/mergedLibrary/*') from ch_rm_orphan_flagstat_mqc.collect{it[1]}
file ('alignment/mergedLibrary/*') from ch_rm_orphan_stats_mqc.collect()
file ('alignment/mergedLibrary/picard_metrics/*') from ch_merge_bam_metrics_mqc.collect()
file ('alignment/mergedLibrary/picard_metrics/*') from ch_collectmetrics_mqc.collect()
file ('macs/*') from ch_macs_mqc.collect().ifEmpty([])
file ('macs/*') from ch_macs_qc_mqc.collect().ifEmpty([])
file ('macs/consensus/*') from ch_macs_consensus_counts_mqc.collect().ifEmpty([])
file ('macs/consensus/*') from ch_macs_consensus_deseq_mqc.collect().ifEmpty([])
file ('preseq/*') from ch_preseq_results.collect().ifEmpty([])
file ('deeptools/*') from ch_plotfingerprint_mqc.collect().ifEmpty([])
file ('deeptools/*') from ch_plotprofile_mqc.collect().ifEmpty([])
file ('phantompeakqualtools/*') from ch_spp_out_mqc.collect().ifEmpty([])
file ('phantompeakqualtools/*') from ch_spp_csv_mqc.collect().ifEmpty([])
file ('software_versions/*') from ch_software_versions_mqc.collect()
file ('workflow_summary/*') from create_workflow_summary(summary)
output:
file "*multiqc_report.html" into ch_multiqc_report
file "*_data"
file "multiqc_plots"
script:
rtitle = custom_runName ? "--title \"$custom_runName\"" : ''
rfilename = custom_runName ? "--filename " + custom_runName.replaceAll('\\W','_').replaceAll('_+','_') + "_multiqc_report" : ''
mqcstats = params.skipMultiQCStats ? '--cl_config "skip_generalstats: true"' : ''
"""
multiqc . -f $rtitle $rfilename --config $multiqc_config \\
-m custom_content -m fastqc -m cutadapt -m samtools -m picard -m preseq -m featureCounts -m deeptools -m phantompeakqualtools \\
$mqcstats
"""
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- REPORTS/DOCUMENTATION -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/*
* STEP 10 - Output description HTML
*/
process output_documentation {
publishDir "${params.outdir}/Documentation", mode: 'copy'
input:
file output_docs from ch_output_docs
output:
file "results_description.html"
script:
"""
markdown_to_html.r $output_docs results_description.html
"""
}
/*
* Completion e-mail notification
*/
workflow.onComplete {
// Set up the e-mail variables
def subject = "[nf-core/chipseq] Successful: $workflow.runName"
if(!workflow.success){
subject = "[nf-core/chipseq] FAILED: $workflow.runName"
}
def email_fields = [:]
email_fields['version'] = workflow.manifest.version
email_fields['runName'] = custom_runName ?: workflow.runName
email_fields['success'] = workflow.success
email_fields['dateComplete'] = workflow.complete
email_fields['duration'] = workflow.duration
email_fields['exitStatus'] = workflow.exitStatus
email_fields['errorMessage'] = (workflow.errorMessage ?: 'None')
email_fields['errorReport'] = (workflow.errorReport ?: 'None')
email_fields['commandLine'] = workflow.commandLine
email_fields['projectDir'] = workflow.projectDir
email_fields['summary'] = summary
email_fields['summary']['Date Started'] = workflow.start
email_fields['summary']['Date Completed'] = workflow.complete
email_fields['summary']['Pipeline script file path'] = workflow.scriptFile
email_fields['summary']['Pipeline script hash ID'] = workflow.scriptId
if(workflow.repository) email_fields['summary']['Pipeline repository Git URL'] = workflow.repository
if(workflow.commitId) email_fields['summary']['Pipeline repository Git Commit'] = workflow.commitId
if(workflow.revision) email_fields['summary']['Pipeline Git branch/tag'] = workflow.revision
if(workflow.container) email_fields['summary']['Docker image'] = workflow.container
email_fields['summary']['Nextflow Version'] = workflow.nextflow.version
email_fields['summary']['Nextflow Build'] = workflow.nextflow.build
email_fields['summary']['Nextflow Compile Timestamp'] = workflow.nextflow.timestamp
// On success try attach the multiqc report
def mqc_report = null
try {
if (workflow.success) {
mqc_report = ch_multiqc_report.getVal()
if (mqc_report.getClass() == ArrayList){
log.warn "[nf-core/chipseq] Found multiple reports from process 'multiqc', will use only one"
mqc_report = mqc_report[0]
}
}
} catch (all) {
log.warn "[nf-core/chipseq] Could not attach MultiQC report to summary email"
}
// Render the TXT template
def engine = new groovy.text.GStringTemplateEngine()
def tf = new File("$baseDir/assets/email_template.txt")
def txt_template = engine.createTemplate(tf).make(email_fields)
def email_txt = txt_template.toString()
// Render the HTML template
def hf = new File("$baseDir/assets/email_template.html")
def html_template = engine.createTemplate(hf).make(email_fields)
def email_html = html_template.toString()
// Render the sendmail template
def smail_fields = [ email: params.email, subject: subject, email_txt: email_txt, email_html: email_html, baseDir: "$baseDir", mqcFile: mqc_report, mqcMaxSize: params.maxMultiqcEmailFileSize.toBytes() ]
def sf = new File("$baseDir/assets/sendmail_template.txt")
def sendmail_template = engine.createTemplate(sf).make(smail_fields)
def sendmail_html = sendmail_template.toString()
// Send the HTML e-mail
if (params.email) {
try {
if( params.plaintext_email ){ throw GroovyException('Send plaintext e-mail, not HTML') }
// Try to send HTML e-mail using sendmail
[ 'sendmail', '-t' ].execute() << sendmail_html
log.info "[nf-core/chipseq] Sent summary e-mail to $params.email (sendmail)"
} catch (all) {
// Catch failures and try with plaintext
[ 'mail', '-s', subject, params.email ].execute() << email_txt
log.info "[nf-core/chipseq] Sent summary e-mail to $params.email (mail)"
}
}
// Write summary e-mail HTML to a file
def output_d = new File( "${params.outdir}/pipeline_info/" )
if( !output_d.exists() ) {
output_d.mkdirs()
}
def output_hf = new File( output_d, "pipeline_report.html" )
output_hf.withWriter { w -> w << email_html }
def output_tf = new File( output_d, "pipeline_report.txt" )
output_tf.withWriter { w -> w << email_txt }
c_reset = params.monochrome_logs ? '' : "\033[0m";
c_purple = params.monochrome_logs ? '' : "\033[0;35m";
c_green = params.monochrome_logs ? '' : "\033[0;32m";
c_red = params.monochrome_logs ? '' : "\033[0;31m";
if (workflow.stats.ignoredCount > 0 && workflow.success) {
log.info "${c_purple}Warning, pipeline completed, but with errored process(es) ${c_reset}"
log.info "${c_red}Number of ignored errored process(es) : ${workflow.stats.ignoredCount} ${c_reset}"
log.info "${c_green}Number of successfully ran process(es) : ${workflow.stats.succeedCount} ${c_reset}"
}
if(workflow.success){
log.info "${c_purple}[nf-core/chipseq]${c_green} Pipeline completed successfully${c_reset}"
} else {
checkHostname()
log.info "${c_purple}[nf-core/chipseq]${c_red} Pipeline completed with errors${c_reset}"
}
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- NF-CORE HEADER -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
def nfcoreHeader(){
// Log colors ANSI codes
c_reset = params.monochrome_logs ? '' : "\033[0m";
c_dim = params.monochrome_logs ? '' : "\033[2m";
c_black = params.monochrome_logs ? '' : "\033[0;30m";
c_green = params.monochrome_logs ? '' : "\033[0;32m";
c_yellow = params.monochrome_logs ? '' : "\033[0;33m";
c_blue = params.monochrome_logs ? '' : "\033[0;34m";
c_purple = params.monochrome_logs ? '' : "\033[0;35m";
c_cyan = params.monochrome_logs ? '' : "\033[0;36m";
c_white = params.monochrome_logs ? '' : "\033[0;37m";
return """ ${c_dim}----------------------------------------------------${c_reset}
${c_green},--.${c_black}/${c_green},-.${c_reset}
${c_blue} ___ __ __ __ ___ ${c_green}/,-._.--~\'${c_reset}
${c_blue} |\\ | |__ __ / ` / \\ |__) |__ ${c_yellow}} {${c_reset}
${c_blue} | \\| | \\__, \\__/ | \\ |___ ${c_green}\\`-._,-`-,${c_reset}
${c_green}`._,._,\'${c_reset}
${c_purple} nf-core/chipseq v${workflow.manifest.version}${c_reset}
${c_dim}----------------------------------------------------${c_reset}
""".stripIndent()
}
def checkHostname(){
def c_reset = params.monochrome_logs ? '' : "\033[0m"
def c_white = params.monochrome_logs ? '' : "\033[0;37m"
def c_red = params.monochrome_logs ? '' : "\033[1;91m"
def c_yellow_bold = params.monochrome_logs ? '' : "\033[1;93m"
if(params.hostnames){
def hostname = "hostname".execute().text.trim()
params.hostnames.each { prof, hnames ->
hnames.each { hname ->
if(hostname.contains(hname) && !workflow.profile.contains(prof)){
log.error "====================================================\n" +
" ${c_red}WARNING!${c_reset} You are running with `-profile $workflow.profile`\n" +
" but your machine hostname is ${c_white}'$hostname'${c_reset}\n" +
" ${c_yellow_bold}It's highly recommended that you use `-profile $prof${c_reset}`\n" +
"============================================================"
}
}
}
}
}
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////
/* -- -- */
/* -- END OF PIPELINE -- */
/* -- -- */
///////////////////////////////////////////////////////////////////////////////
///////////////////////////////////////////////////////////////////////////////Standardised workflows
monolithic
Breaking down monoliths ...





... into reusable modular components
How to go about it?
Need to change often
Siloed tool containers
Don't do much by themselves
Monolithic pipelines

The solution
The premise:
Workflow based development
Component based development
Components are modular pieces with some basic rules:

Component A
- Input/Output
- Parameters
- Resources
Component B

- Input/Output
- Parameters
- Resources
Flowcraft - Easy way to create & glue lego components
With this framework, building workflows becomes dead simple:
flowcraft build -t 'trimmomatic fastqc spades pilon' -o my_nextflow_pipeline
Results in the following workflow DAG (direct acyclic graph)
It's easy to get experimental:
flowcraft build -t 'trimmomatic fastqc skesa pilon' -o my_nextflow_pipeline
Switch spades for skesa
It's easy to get wild:
flowcraft build -t 'reads_download (
spades | skesa pilon (abricate | chewbbaca) | megahit |
fastqc_trimmomatic fastqc (spades pilon (
mlst | prokka | chewbbaca) | skesa pilon abricate))' -o my_nextflow_pipelinewait, what?
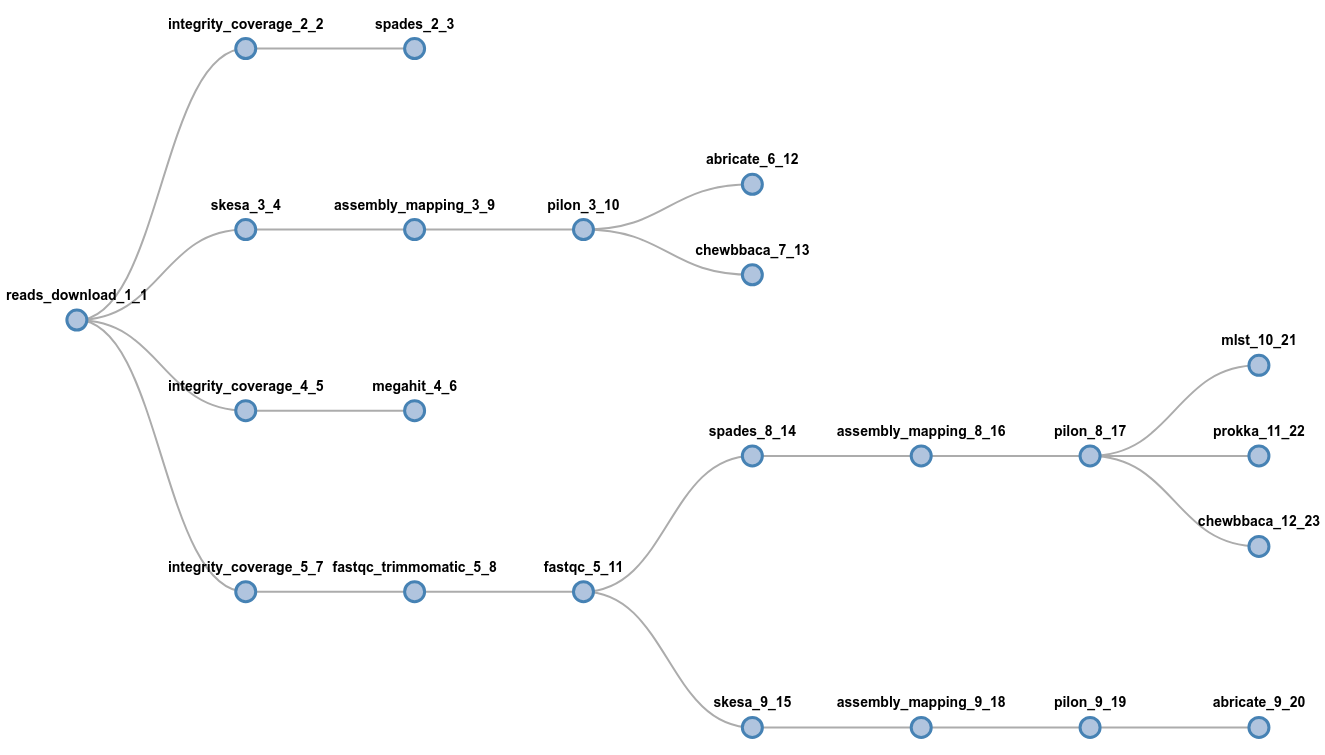
Flowcraft - Easy way to create & glue lego components
Flowcraft - Easy way to create & glue lego components
#!/usr/bin/env nextflow
import Helper
import CollectInitialMetadata
// Pipeline version
if (workflow.commitId){
version = "0.1 $workflow.revision"
} else {
version = "0.1 (local version)"
}
params.help = false
if (params.help){
Help.print_help(params)
exit 0
}
def infoMap = [:]
if (params.containsKey("fastq")){
infoMap.put("fastq", file(params.fastq).size())
}
if (params.containsKey("fasta")){
if (file(params.fasta) instanceof LinkedList){
infoMap.put("fasta", file(params.fasta).size())
} else {
infoMap.put("fasta", 1)
}
}
if (params.containsKey("accessions")){
// checks if params.accessions is different from null
if (params.accessions) {
BufferedReader reader = new BufferedReader(new FileReader(params.accessions));
int lines = 0;
while (reader.readLine() != null) lines++;
reader.close();
infoMap.put("accessions", lines)
}
}
Help.start_info(infoMap, "$workflow.start", "$workflow.profile")
CollectInitialMetadata.print_metadata(workflow)
// Placeholder for main input channels
if (!params.accessions){ exit 1, "'accessions' parameter missing" }
IN_accessions_raw = Channel.fromPath(params.accessions).ifEmpty { exit 1, "No accessions file provided with path:'${params.accessions}'" }
// Placeholder for secondary input channels
// Placeholder for extra input channels
// Placeholder to fork the raw input channel
IN_accessions_raw.set{ reads_download_in_1_0 }
if (params.asperaKey_1_1){
if (file(params.asperaKey_1_1).exists()){
IN_asperaKey_1_1 = Channel.fromPath(params.asperaKey_1_1)
} else {
IN_asperaKey_1_1 = Channel.value("")
}
} else {
IN_asperaKey_1_1 = Channel.value("")
}
process reads_download_1_1 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 1_1 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 1_1 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 1_1 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId reads_download_1_1 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { accession_id }
publishDir "reads", pattern: "${accession_id}/*fq.gz"
maxRetries 1
input:
set val(accession_id), val(name) from reads_download_in_1_0.splitText(){ it.trim() }.unique().filter{ it != "" }.map{ it.split().length > 1 ? ["accession": it.split()[0], "name": it.split()[1]] : [it.split()[0], null] }
each file(aspera_key) from IN_asperaKey_1_1
output:
set val({ "$name" != "null" ? "$name" : "$accession_id" }), file("${accession_id}/*fq.gz") optional true into _reads_download_out_1_0
set accession_id, val("1_1_reads_download"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_reads_download_1_1
set accession_id, val("reads_download_1_1"), val("1_1"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_reads_download_1_1
file ".versions"
script:
"""
{
# getSeqENA requires accession numbers to be provided as a text file
echo "${accession_id}" >> accession_file.txt
# Set default status value. It will be overwritten if anything goes wrong
echo "pass" > ".status"
if [ -f $aspera_key ]; then
asperaOpt="-a $aspera_key"
else
asperaOpt=""
fi
getSeqENA.py -l accession_file.txt \$asperaOpt -o ./ --SRAopt --downloadCramBam
# If a name has been provided along with the accession, rename the
# fastq files.
if [ $name != null ];
then
echo renaming pattern '${accession_id}' to '${name}' && cd ${accession_id} && rename "s/${accession_id}/${name}/" *.gz
fi
} || {
# If exit code other than 0
if [ \$? -eq 0 ]
then
echo "pass" > .status
else
echo "fail" > .status
echo "Could not download accession $accession_id" > .fail
fi
}
version_str="{'version':[{'program':'getSeqENA.py','version':'1.3'}]}"
echo \$version_str > .versions
"""
}
_reads_download_out_1_0.into{ reads_download_out_1_0;spades_in_1_1;skesa_in_1_2;megahit_in_1_3;fastqc_trimmomatic_in_1_4;_LAST_fastq_3_9 }
IN_genome_size_2_2 = Channel.value(params.genomeSize_2_2)
.map{it -> it.toString().isNumber() ? it : exit(1, "The genomeSize parameter must be a number or a float. Provided value: '${params.genomeSize__2_2}'")}
IN_min_coverage_2_2 = Channel.value(params.minCoverage_2_2)
.map{it -> it.toString().isNumber() ? it : exit(1, "The minCoverage parameter must be a number or a float. Provided value: '${params.minCoverage__2_2}'")}
process integrity_coverage_2_2 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 2_2 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 2_2 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 2_2 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId integrity_coverage_2_2 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(fastq_pair) from spades_in_1_1
val gsize from IN_genome_size_2_2
val cov from IN_min_coverage_2_2
// This channel is for the custom options of the integrity_coverage.py
// script. See the script's documentation for more information.
val opts from Channel.value('')
output:
set sample_id,
file(fastq_pair),
file('*_encoding'),
file('*_phred'),
file('*_coverage'),
file('*_max_len') into MAIN_integrity_2_2
file('*_report') optional true into LOG_report_coverage1_2_2
set sample_id, val("2_2_integrity_coverage"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_integrity_coverage_2_2
set sample_id, val("integrity_coverage_2_2"), val("2_2"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_integrity_coverage_2_2
file ".versions"
script:
template "integrity_coverage.py"
}
// TRIAGE OF CORRUPTED SAMPLES
LOG_corrupted_2_2 = Channel.create()
MAIN_PreCoverageCheck_2_2 = Channel.create()
// Corrupted samples have the 2nd value with 'corrupt'
MAIN_integrity_2_2.choice(LOG_corrupted_2_2, MAIN_PreCoverageCheck_2_2) {
a -> a[2].text == "corrupt" ? 0 : 1
}
// TRIAGE OF LOW COVERAGE SAMPLES
integrity_coverage_out_2_1_dep = Channel.create()
SIDE_phred_2_2 = Channel.create()
SIDE_max_len_2_2 = Channel.create()
MAIN_PreCoverageCheck_2_2
// Low coverage samples have the 4th value of the Channel with 'fail'
.filter{ it[4].text != "fail" }
// For the channel to proceed with FastQ in 'sample_good' and the
// Phred scores for each sample in 'SIDE_phred'
.separate(integrity_coverage_out_2_1_dep, SIDE_phred_2_2, SIDE_max_len_2_2){
a -> [ [a[0], a[1]], [a[0], a[3].text], [a[0], a[5].text] ]
}
/** REPORT_COVERAGE - PLUG-IN
This process will report the expected coverage for each non-corrupted sample
and write the results to 'reports/coverage/estimated_coverage_initial.csv'
*/
process report_coverage_2_2 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/coverage_2_2/'
input:
file(report) from LOG_report_coverage1_2_2.filter{ it.text != "corrupt" }.collect()
output:
file 'estimated_coverage_initial.csv'
"""
echo Sample,Estimated coverage,Status >> estimated_coverage_initial.csv
cat $report >> estimated_coverage_initial.csv
"""
}
/** REPORT_CORRUPT - PLUG-IN
This process will report the corrupted samples and write the results to
'reports/corrupted/corrupted_samples.txt'
*/
process report_corrupt_2_2 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/corrupted_2_2/'
input:
val sample_id from LOG_corrupted_2_2.collect{it[0]}
output:
file 'corrupted_samples.txt'
"""
echo ${sample_id.join(",")} | tr "," "\n" >> corrupted_samples.txt
"""
}
SIDE_max_len_2_2.set{ SIDE_max_len_2_3 }
if ( !params.spadesMinCoverage_2_3.toString().isNumber() ){
exit 1, "'spadesMinCoverage_2_3' parameter must be a number. Provided value: '${params.spadesMinCoverage_2_3}'"
}
if ( !params.spadesMinKmerCoverage_2_3.toString().isNumber()){
exit 1, "'spadesMinKmerCoverage_2_3' parameter must be a number. Provided value: '${params.spadesMinKmerCoverage_2_3}'"
}
IN_spades_opts_2_3 = Channel.value(
[params.spadesMinCoverage_2_3,
params.spadesMinKmerCoverage_2_3
])
if ( params.spadesKmers_2_3.toString().split(" ").size() <= 1 ){
if (params.spadesKmers_2_3.toString() != 'auto'){
exit 1, "'spadesKmers_2_3' parameter must be a sequence of space separated numbers or 'auto'. Provided value: ${params.spadesKmers_2_3}"
}
}
IN_spades_kmers_2_3 = Channel.value(params.spadesKmers_2_3)
clear = params.clearInput_2_3 ? "true" : "false"
disable_rr = params.disableRR_2_3 ? "true" : "false"
checkpointClear_2_3 = Channel.value(clear)
disableRR_2_3 = Channel.value(disable_rr)
process spades_2_3 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 2_3 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 2_3 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 2_3 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId spades_2_3 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir 'results/assembly/spades_2_3/', pattern: '*_spades*.fasta', mode: 'copy'
publishDir "results/assembly/spades_2_3/$sample_id", pattern: "*.gfa", mode: "copy"
publishDir "results/assembly/spades_2_3/$sample_id", pattern: "*.fastg", mode: "copy"
input:
set sample_id, file(fastq_pair), max_len from integrity_coverage_out_2_1_dep.join(SIDE_max_len_2_3)
val opts from IN_spades_opts_2_3
val kmers from IN_spades_kmers_2_3
val clear from checkpointClear_2_3
val disable_rr from disableRR_2_3
output:
set sample_id, file('*_spades*.fasta') into spades_out_2_1
file "*.fastg" optional true
file "*.gfa" into gfa1_2_3
set sample_id, val("2_3_spades"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_spades_2_3
set sample_id, val("spades_2_3"), val("2_3"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_spades_2_3
file ".versions"
script:
template "spades.py"
}
clear = params.clearInput_3_4 ? "true" : "false"
checkpointClear_3_4 = Channel.value(clear)
process skesa_3_4 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 3_4 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 3_4 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 3_4 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId skesa_3_4 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir 'results/assembly/skesa_3_4', pattern: '*skesa*.fasta', mode: 'copy'
input:
set sample_id, file(fastq_pair) from skesa_in_1_2
val clear from checkpointClear_3_4
output:
set sample_id, file('*.fasta') into skesa_out_3_2
set sample_id, val("3_4_skesa"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_skesa_3_4
set sample_id, val("skesa_3_4"), val("3_4"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_skesa_3_4
file ".versions"
script:
template "skesa.py"
}
IN_genome_size_4_5 = Channel.value(params.genomeSize_4_5)
.map{it -> it.toString().isNumber() ? it : exit(1, "The genomeSize parameter must be a number or a float. Provided value: '${params.genomeSize__4_5}'")}
IN_min_coverage_4_5 = Channel.value(params.minCoverage_4_5)
.map{it -> it.toString().isNumber() ? it : exit(1, "The minCoverage parameter must be a number or a float. Provided value: '${params.minCoverage__4_5}'")}
process integrity_coverage_4_5 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 4_5 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 4_5 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 4_5 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId integrity_coverage_4_5 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(fastq_pair) from megahit_in_1_3
val gsize from IN_genome_size_4_5
val cov from IN_min_coverage_4_5
// This channel is for the custom options of the integrity_coverage.py
// script. See the script's documentation for more information.
val opts from Channel.value('')
output:
set sample_id,
file(fastq_pair),
file('*_encoding'),
file('*_phred'),
file('*_coverage'),
file('*_max_len') into MAIN_integrity_4_5
file('*_report') optional true into LOG_report_coverage1_4_5
set sample_id, val("4_5_integrity_coverage"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_integrity_coverage_4_5
set sample_id, val("integrity_coverage_4_5"), val("4_5"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_integrity_coverage_4_5
file ".versions"
script:
template "integrity_coverage.py"
}
// TRIAGE OF CORRUPTED SAMPLES
LOG_corrupted_4_5 = Channel.create()
MAIN_PreCoverageCheck_4_5 = Channel.create()
// Corrupted samples have the 2nd value with 'corrupt'
MAIN_integrity_4_5.choice(LOG_corrupted_4_5, MAIN_PreCoverageCheck_4_5) {
a -> a[2].text == "corrupt" ? 0 : 1
}
// TRIAGE OF LOW COVERAGE SAMPLES
integrity_coverage_out_4_3_dep = Channel.create()
SIDE_phred_4_5 = Channel.create()
SIDE_max_len_4_5 = Channel.create()
MAIN_PreCoverageCheck_4_5
// Low coverage samples have the 4th value of the Channel with 'fail'
.filter{ it[4].text != "fail" }
// For the channel to proceed with FastQ in 'sample_good' and the
// Phred scores for each sample in 'SIDE_phred'
.separate(integrity_coverage_out_4_3_dep, SIDE_phred_4_5, SIDE_max_len_4_5){
a -> [ [a[0], a[1]], [a[0], a[3].text], [a[0], a[5].text] ]
}
/** REPORT_COVERAGE - PLUG-IN
This process will report the expected coverage for each non-corrupted sample
and write the results to 'reports/coverage/estimated_coverage_initial.csv'
*/
process report_coverage_4_5 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/coverage_4_5/'
input:
file(report) from LOG_report_coverage1_4_5.filter{ it.text != "corrupt" }.collect()
output:
file 'estimated_coverage_initial.csv'
"""
echo Sample,Estimated coverage,Status >> estimated_coverage_initial.csv
cat $report >> estimated_coverage_initial.csv
"""
}
/** REPORT_CORRUPT - PLUG-IN
This process will report the corrupted samples and write the results to
'reports/corrupted/corrupted_samples.txt'
*/
process report_corrupt_4_5 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/corrupted_4_5/'
input:
val sample_id from LOG_corrupted_4_5.collect{it[0]}
output:
file 'corrupted_samples.txt'
"""
echo ${sample_id.join(",")} | tr "," "\n" >> corrupted_samples.txt
"""
}
SIDE_max_len_4_5.set{ SIDE_max_len_4_6 }
if ( params.megahitKmers_4_6.toString().split(" ").size() <= 1 ){
if (params.megahitKmers_4_6.toString() != 'auto'){
exit 1, "'megahitKmers_4_6' parameter must be a sequence of space separated numbers or 'auto'. Provided value: ${params.megahitKmers_4_6}"
}
}
IN_megahit_kmers_4_6 = Channel.value(params.megahitKmers_4_6)
clear = params.clearInput_4_6 ? "true" : "false"
checkpointClear_4_6 = Channel.value(clear)
process megahit_4_6 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 4_6 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 4_6 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 4_6 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId megahit_4_6 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir 'results/assembly/megahit_4_6/', pattern: '*_megahit*.fasta', mode: 'copy'
input:
set sample_id, file(fastq_pair), max_len from integrity_coverage_out_4_3_dep.join(SIDE_max_len_4_6)
val kmers from IN_megahit_kmers_4_6
val clear from checkpointClear_4_6
output:
set sample_id, file('*megahit*.fasta') into megahit_out_4_3
set sample_id, file('megahit/intermediate_contigs/k*.contigs.fa') into IN_fastg4_6
set sample_id, val("4_6_megahit"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_megahit_4_6
set sample_id, val("megahit_4_6"), val("4_6"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_megahit_4_6
file ".versions"
script:
template "megahit.py"
}
fastg = params.fastg_4_6 ? "true" : "false"
process megahit_fastg_4_6{
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 4_6 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 4_6 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 4_6 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId megahit_4_6 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir "results/assembly/megahit_4_6/$sample_id", pattern: "*.fastg"
input:
set sample_id, file(kmer_files) from IN_fastg4_6
val run_fastg from fastg
output:
file "*.fastg" optional true
set sample_id, val("4_6_megahit_fastg"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_megahit_fastg_4_6
set sample_id, val("megahit_fastg_4_6"), val("4_6"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_megahit_fastg_4_6
file ".versions"
script:
"""
if [ ${run_fastg} == "true" ]
then
for kmer_file in ${kmer_files};
do
echo \$kmer_file
k=\$(echo \$kmer_file | cut -d '.' -f 1);
echo \$k
megahit_toolkit contig2fastg \$k \$kmer_file > \$kmer_file'.fastg';
done
fi
"""
}
IN_genome_size_5_7 = Channel.value(params.genomeSize_5_7)
.map{it -> it.toString().isNumber() ? it : exit(1, "The genomeSize parameter must be a number or a float. Provided value: '${params.genomeSize__5_7}'")}
IN_min_coverage_5_7 = Channel.value(params.minCoverage_5_7)
.map{it -> it.toString().isNumber() ? it : exit(1, "The minCoverage parameter must be a number or a float. Provided value: '${params.minCoverage__5_7}'")}
process integrity_coverage_5_7 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_7 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_7 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_7 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId integrity_coverage_5_7 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(fastq_pair) from fastqc_trimmomatic_in_1_4
val gsize from IN_genome_size_5_7
val cov from IN_min_coverage_5_7
// This channel is for the custom options of the integrity_coverage.py
// script. See the script's documentation for more information.
val opts from Channel.value('')
output:
set sample_id,
file(fastq_pair),
file('*_encoding'),
file('*_phred'),
file('*_coverage'),
file('*_max_len') into MAIN_integrity_5_7
file('*_report') optional true into LOG_report_coverage1_5_7
set sample_id, val("5_7_integrity_coverage"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_integrity_coverage_5_7
set sample_id, val("integrity_coverage_5_7"), val("5_7"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_integrity_coverage_5_7
file ".versions"
script:
template "integrity_coverage.py"
}
// TRIAGE OF CORRUPTED SAMPLES
LOG_corrupted_5_7 = Channel.create()
MAIN_PreCoverageCheck_5_7 = Channel.create()
// Corrupted samples have the 2nd value with 'corrupt'
MAIN_integrity_5_7.choice(LOG_corrupted_5_7, MAIN_PreCoverageCheck_5_7) {
a -> a[2].text == "corrupt" ? 0 : 1
}
// TRIAGE OF LOW COVERAGE SAMPLES
integrity_coverage_out_5_4_dep = Channel.create()
SIDE_phred_5_7 = Channel.create()
SIDE_max_len_5_7 = Channel.create()
MAIN_PreCoverageCheck_5_7
// Low coverage samples have the 4th value of the Channel with 'fail'
.filter{ it[4].text != "fail" }
// For the channel to proceed with FastQ in 'sample_good' and the
// Phred scores for each sample in 'SIDE_phred'
.separate(integrity_coverage_out_5_4_dep, SIDE_phred_5_7, SIDE_max_len_5_7){
a -> [ [a[0], a[1]], [a[0], a[3].text], [a[0], a[5].text] ]
}
/** REPORT_COVERAGE - PLUG-IN
This process will report the expected coverage for each non-corrupted sample
and write the results to 'reports/coverage/estimated_coverage_initial.csv'
*/
process report_coverage_5_7 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/coverage_5_7/'
input:
file(report) from LOG_report_coverage1_5_7.filter{ it.text != "corrupt" }.collect()
output:
file 'estimated_coverage_initial.csv'
"""
echo Sample,Estimated coverage,Status >> estimated_coverage_initial.csv
cat $report >> estimated_coverage_initial.csv
"""
}
/** REPORT_CORRUPT - PLUG-IN
This process will report the corrupted samples and write the results to
'reports/corrupted/corrupted_samples.txt'
*/
process report_corrupt_5_7 {
// This process can only use a single CPU
cpus 1
publishDir 'reports/corrupted_5_7/'
input:
val sample_id from LOG_corrupted_5_7.collect{it[0]}
output:
file 'corrupted_samples.txt'
"""
echo ${sample_id.join(",")} | tr "," "\n" >> corrupted_samples.txt
"""
}
SIDE_phred_5_7.set{ SIDE_phred_5_8 }
SIDE_max_len_5_7.set{ SIDE_max_len_8_14 }
// Check sliding window parameter
if ( params.trimSlidingWindow_5_8.toString().split(":").size() != 2 ){
exit 1, "'trimSlidingWindow_5_8' parameter must contain two values separated by a ':'. Provided value: '${params.trimSlidingWindow_5_8}'"
}
if ( !params.trimLeading_5_8.toString().isNumber() ){
exit 1, "'trimLeading_5_8' parameter must be a number. Provide value: '${params.trimLeading_5_8}'"
}
if ( !params.trimTrailing_5_8.toString().isNumber() ){
exit 1, "'trimTrailing_5_8' parameter must be a number. Provide value: '${params.trimTrailing_5_8}'"
}
if ( !params.trimMinLength_5_8.toString().isNumber() ){
exit 1, "'trimMinLength_5_8' parameter must be a number. Provide value: '${params.trimMinLength_5_8}'"
}
IN_trimmomatic_opts_5_8 = Channel.value([params.trimSlidingWindow_5_8,params.trimLeading_5_8,params.trimTrailing_5_8,params.trimMinLength_5_8])
IN_adapters_5_8 = Channel.value(params.adapters_5_8)
clear = params.clearInput_5_8 ? "true" : "false"
checkpointClear_5_8 = Channel.value(clear)
process fastqc_5_8 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_8 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId fastqc_trimmomatic_5_8 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir "reports/fastqc_5_8/", pattern: "*.html"
input:
set sample_id, file(fastq_pair) from integrity_coverage_out_5_4_dep
val ad from Channel.value('None')
output:
set sample_id, file(fastq_pair), file('pair_1*'), file('pair_2*') into MAIN_fastqc_out_5_8
file "*html"
set sample_id, val("5_8_fastqc"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_fastqc_5_8
set sample_id, val("fastqc_5_8"), val("5_8"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_fastqc_5_8
file ".versions"
script:
template "fastqc.py"
}
/** FASTQC_REPORT - MAIN
This process will parse the result files from a FastQC analyses and output
the optimal_trim information for Trimmomatic
*/
process fastqc_report_5_8 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_8 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId fastqc_trimmomatic_5_8 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
publishDir 'reports/fastqc_5_8/run_1/', pattern: '*summary.txt', mode: 'copy'
input:
set sample_id, file(fastq_pair), file(result_p1), file(result_p2) from MAIN_fastqc_out_5_8
val opts from Channel.value("--ignore-tests")
output:
set sample_id, file(fastq_pair), 'optimal_trim', ".status" into _MAIN_fastqc_trim_5_8
file '*_trim_report' into LOG_trim_5_8
file "*_status_report" into LOG_fastqc_report_5_8
file "${sample_id}_*_summary.txt" optional true
set sample_id, val("5_8_fastqc_report"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_fastqc_report_5_8
set sample_id, val("fastqc_report_5_8"), val("5_8"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_fastqc_report_5_8
file ".versions"
script:
template "fastqc_report.py"
}
MAIN_fastqc_trim_5_8 = Channel.create()
_MAIN_fastqc_trim_5_8
.filter{ it[3].text == "pass" }
.map{ [it[0], it[1], file(it[2]).text] }
.into(MAIN_fastqc_trim_5_8)
/** TRIM_REPORT - PLUG-IN
This will collect the optimal trim points assessed by the fastqc_report
process and write the results of all samples in a single csv file
*/
process trim_report_5_8 {
publishDir 'reports/fastqc_5_8/', mode: 'copy'
input:
file trim from LOG_trim_5_8.collect()
output:
file "FastQC_trim_report.csv"
"""
echo Sample,Trim begin, Trim end >> FastQC_trim_report.csv
cat $trim >> FastQC_trim_report.csv
"""
}
process compile_fastqc_status_5_8 {
publishDir 'reports/fastqc_5_8/', mode: 'copy'
input:
file rep from LOG_fastqc_report_5_8.collect()
output:
file 'FastQC_1run_report.csv'
"""
echo Sample, Failed? >> FastQC_1run_report.csv
cat $rep >> FastQC_1run_report.csv
"""
}
/** TRIMMOMATIC - MAIN
This process will execute trimmomatic. Currently, the main channel requires
information on the trim_range and phred score.
*/
process trimmomatic_5_8 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_8 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_8 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId fastqc_trimmomatic_5_8 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir "results/trimmomatic_5_8", pattern: "*.gz"
input:
set sample_id, file(fastq_pair), trim_range, phred from MAIN_fastqc_trim_5_8.join(SIDE_phred_5_8)
val opts from IN_trimmomatic_opts_5_8
val ad from IN_adapters_5_8
val clear from checkpointClear_5_8
output:
set sample_id, "${sample_id}_*trim.fastq.gz" into fastqc_trimmomatic_out_5_4
file 'trimmomatic_report.csv'
set sample_id, val("5_8_trimmomatic"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_trimmomatic_5_8
set sample_id, val("trimmomatic_5_8"), val("5_8"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_trimmomatic_5_8
file ".versions"
script:
template "trimmomatic.py"
}
if ( !params.minAssemblyCoverage_3_9.toString().isNumber() ){
if (params.minAssemblyCoverage_3_9.toString() != 'auto'){
exit 1, "'minAssemblyCoverage_3_9' parameter must be a number or 'auto'. Provided value: ${params.minAssemblyCoverage_3_9}"
}
}
if ( !params.AMaxContigs_3_9.toString().isNumber() ){
exit 1, "'AMaxContigs_3_9' parameter must be a number. Provide value: '${params.AMaxContigs_3_9}'"
}
IN_assembly_mapping_opts_3_9 = Channel.value([params.minAssemblyCoverage_3_9,params.AMaxContigs_3_9])
IN_genome_size_3_9 = Channel.value(params.genomeSize_3_9)
process assembly_mapping_3_9 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 3_9 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 3_9 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 3_9 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_3_9 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(fastq) from skesa_out_3_2.join(_LAST_fastq_3_9)
output:
set sample_id, file(assembly), 'coverages.tsv', 'coverage_per_bp.tsv', 'sorted.bam', 'sorted.bam.bai' into MAIN_am_out_3_9
set sample_id, file("coverage_per_bp.tsv") optional true into SIDE_BpCoverage_3_9
set sample_id, val("3_9_assembly_mapping"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_assembly_mapping_3_9
set sample_id, val("assembly_mapping_3_9"), val("3_9"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_assembly_mapping_3_9
file ".versions"
script:
"""
{
echo [DEBUG] BUILDING BOWTIE INDEX FOR ASSEMBLY: $assembly >> .command.log 2>&1
bowtie2-build --threads ${task.cpus} $assembly genome_index >> .command.log 2>&1
echo [DEBUG] MAPPING READS FROM $fastq >> .command.log 2>&1
bowtie2 -q --very-sensitive-local --threads ${task.cpus} -x genome_index -1 ${fastq[0]} -2 ${fastq[1]} -S mapping.sam >> .command.log 2>&1
echo [DEBUG] CONVERTING AND SORTING SAM TO BAM >> .command.log 2>&1
samtools sort -o sorted.bam -O bam -@ ${task.cpus} mapping.sam && rm *.sam >> .command.log 2>&1
echo [DEBUG] CREATING BAM INDEX >> .command.log 2>&1
samtools index sorted.bam >> .command.log 2>&1
echo [DEBUG] ESTIMATING READ DEPTH >> .command.log 2>&1
parallel -j ${task.cpus} samtools depth -ar {} sorted.bam \\> {}.tab ::: \$(grep ">" $assembly | cut -c 2- | tr " " "_")
# Insert 0 coverage count in empty files. See Issue #2
echo [DEBUG] REMOVING EMPTY FILES >> .command.log 2>&1
find . -size 0 -print0 | xargs -0 -I{} sh -c 'echo -e 0"\t"0"\t"0 > "{}"'
echo [DEBUG] COMPILING COVERAGE REPORT >> .command.log 2>&1
parallel -j ${task.cpus} echo -n {.} '"\t"' '&&' cut -f3 {} '|' paste -sd+ '|' bc >> coverages.tsv ::: *.tab
cat *.tab > coverage_per_bp.tsv
rm *.tab
if [ -f "coverages.tsv" ]
then
echo pass > .status
else
echo fail > .status
fi
echo -n "" > .report.json
echo -n "" > .versions
} || {
echo fail > .status
}
"""
}
/** PROCESS_ASSEMBLY_MAPPING - MAIN
Processes the results from the assembly_mapping process and filters the
assembly contigs based on coverage and length thresholds.
*/
process process_assembly_mapping_3_9 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 3_9 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 3_9 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 3_9 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_3_9 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(assembly), file(coverage), file(coverage_bp), file(bam_file), file(bam_index) from MAIN_am_out_3_9
val opts from IN_assembly_mapping_opts_3_9
val gsize from IN_genome_size_3_9
output:
set sample_id, '*_filt.fasta', 'filtered.bam', 'filtered.bam.bai' into assembly_mapping_out_3_5_dep
set sample_id, val("3_9_process_am"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_process_am_3_9
set sample_id, val("process_am_3_9"), val("3_9"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_process_am_3_9
file ".versions"
script:
template "process_assembly_mapping.py"
}
SIDE_BpCoverage_3_9.set{ SIDE_BpCoverage_3_10 }
clear = params.clearInput_3_10 ? "true" : "false"
checkpointClear_3_10 = Channel.value(clear)
process pilon_3_10 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 3_10 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 3_10 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 3_10 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_3_10 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
echo false
publishDir 'results/assembly/pilon_3_10/', mode: 'copy', pattern: "*.fasta"
input:
set sample_id, file(assembly), file(bam_file), file(bam_index) from assembly_mapping_out_3_5_dep
val clear from checkpointClear_3_10
output:
set sample_id, '*_polished.fasta' into _pilon_out_3_5, pilon_report_3_10
set sample_id, val("3_10_pilon"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_3_10
set sample_id, val("pilon_3_10"), val("3_10"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_3_10
file ".versions"
script:
"""
{
pilon_mem=${String.valueOf(task.memory).substring(0, String.valueOf(task.memory).length() - 1).replaceAll("\\s", "")}
java -jar -Xms256m -Xmx\${pilon_mem} /NGStools/pilon-1.22.jar --genome $assembly --frags $bam_file --output ${assembly.name.replaceFirst(~/\.[^\.]+$/, '')}_polished --changes --threads $task.cpus >> .command.log 2>&1
echo pass > .status
if [ "$clear" = "true" ];
then
work_regex=".*/work/.{2}/.{30}/.*"
assembly_file=\$(readlink -f \$(pwd)/${assembly})
bam_file=\$(readlink -f \$(pwd)/${bam_file})
if [[ "\$assembly_file" =~ \$work_regex ]]; then
rm \$assembly_file \$bam_file
fi
fi
} || {
echo fail > .status
}
"""
}
process pilon_report_3_10 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
afterScript "report_POST.sh $params.projectId $params.pipelineId 3_10 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_3_10 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(coverage_bp) from pilon_report_3_10.join(SIDE_BpCoverage_3_10)
output:
file "*_assembly_report.csv" into pilon_report_out_3_10
set sample_id, val("3_10_pilon_report"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_report_3_10
set sample_id, val("pilon_report_3_10"), val("3_10"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_report_3_10
file ".versions"
script:
template "assembly_report.py"
}
process compile_pilon_report_3_10 {
publishDir "reports/assembly/pilon_3_10/", mode: 'copy'
input:
file(report) from pilon_report_out_3_10.collect()
output:
file "pilon_assembly_report.csv"
"""
echo Sample,Number of contigs,Average contig size,N50,Total assembly length,GC content,Missing data > pilon_assembly_report.csv
cat $report >> pilon_assembly_report.csv
"""
}
_pilon_out_3_5.into{ pilon_out_3_5;abricate_in_3_7;chewbbaca_in_3_8 }
IN_adapters_5_11 = Channel.value(params.adapters_5_11)
process fastqc2_5_11 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_11 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_11 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_11 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId fastqc_5_11 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir "reports/fastqc_5_11/", pattern: "*.html"
input:
set sample_id, file(fastq_pair) from fastqc_trimmomatic_out_5_4
val ad from IN_adapters_5_11
output:
set sample_id, file(fastq_pair), file('pair_1*'), file('pair_2*') into MAIN_fastqc_out_5_11
file "*html"
set sample_id, val("5_11_fastqc2"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_fastqc2_5_11
set sample_id, val("fastqc2_5_11"), val("5_11"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_fastqc2_5_11
file ".versions"
script:
template "fastqc.py"
}
process fastqc2_report_5_11 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 5_11 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 5_11 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 5_11 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId fastqc_5_11 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
publishDir 'reports/fastqc_5_11/run_2/', pattern: '*summary.txt', mode: 'copy'
input:
set sample_id, file(fastq_pair), file(result_p1), file(result_p2) from MAIN_fastqc_out_5_11
val opts from Channel.value("")
output:
set sample_id, file(fastq_pair), '.status' into MAIN_fastqc_report_5_11
file "*_status_report" into LOG_fastqc_report_5_11
file "${sample_id}_*_summary.txt" optional true
set sample_id, val("5_11_fastqc2_report"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_fastqc2_report_5_11
set sample_id, val("fastqc2_report_5_11"), val("5_11"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_fastqc2_report_5_11
file ".versions"
script:
template "fastqc_report.py"
}
process compile_fastqc_status2_5_11 {
publishDir 'reports/fastqc_5_11/', mode: 'copy'
input:
file rep from LOG_fastqc_report_5_11.collect()
output:
file 'FastQC_2run_report.csv'
"""
echo Sample, Failed? >> FastQC_2run_report.csv
cat $rep >> FastQC_2run_report.csv
"""
}
_fastqc_out_5_6 = Channel.create()
MAIN_fastqc_report_5_11
.filter{ it[2].text == "pass" }
.map{ [it[0], it[1]] }
.into(_fastqc_out_5_6)
_fastqc_out_5_6.into{ fastqc_out_5_6;spades_in_5_9;skesa_in_5_10;_LAST_fastq_8_16;_LAST_fastq_9_18 }
if ( params.abricateDataDir_6_12 ){
if ( !file(params.abricateDataDir_6_12).exists() ){
exit 1, "'abricateDataDir_6_12' data directory was not found: '${params.abricateDatabases_6_12}'"
}
dataDirOpt = "--datadir ${params.abricateDataDir_6_12}"
} else {
dataDirOpt = ""
}
if ( !params.abricateMinId_6_12.toString().isNumber() ){
exit 1, "'abricateMinId_6_12' parameter must be a number. Provide value: '${params.abricateMinId_6_12}'"
}
if ( !params.abricateMinCov_6_12.toString().isNumber() ){
exit 1, "'abricateMinCov_6_12' parameter must be a number. Provide value: '${params.abricateMinCov_6_12}'"
}
process abricate_6_12 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 6_12 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 6_12 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 6_12 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId abricate_6_12 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { "${sample_id} ${db}" }
publishDir "results/annotation/abricate_6_12/${sample_id}"
input:
set sample_id, file(assembly) from abricate_in_3_7
each db from params.abricateDatabases_6_12
val min_id from Channel.value(params.abricateMinId_6_12)
val min_cov from Channel.value(params.abricateMinCov_6_12)
output:
file '*.tsv' into abricate_out_6_12
set sample_id, val("6_12_abricate_$db"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_abricate_6_12
set sample_id, val("abricate_6_12_$db"), val("6_12"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_abricate_6_12
file ".versions"
script:
"""
{
# Run abricate
abricate $dataDirOpt --minid $min_id --mincov $min_cov --db $db $assembly > ${sample_id}_abr_${db}.tsv
echo pass > .status
} || {
echo fail > .status
}
"""
}
process process_abricate_6_12 {
tag "process_abricate_6_12"
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
afterScript "report_POST.sh $params.projectId $params.pipelineId 6_12 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId abricate_6_12 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
}
input:
file abricate_file from abricate_out_6_12.collect()
output:
set val('process_abricate'), val("6_12_process_abricate"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_process_abricate_6_12
set val('process_abricate'), val("process_abricate_6_12"), val("6_12"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_process_abricate_6_12
file ".versions"
script:
template "process_abricate.py"
}
if ( !params.schemaPath_7_13 ){
exit 1, "'schemaPath_7_13' parameter missing"
}
if ( params.chewbbacaTraining_7_13){
if (!file(params.chewbbacaTraining_7_13).exists()) {
exit 1, "'chewbbacaTraining_7_13' file was not found: '${params.chewbbacaTraining_7_13}'"
}
}
if ( params.schemaSelectedLoci_7_13){
if (!file(params.schemaSelectedLoci_7_13).exists()) {
exit 1, "'schemaSelectedLoci_7_13' file was not found: '${params.schemaSelectedLoci_7_13}'"
}
}
if ( params.schemaCore_7_13){
if (!file(params.schemaCore_7_13).exists()) {
exit 1, "'schemaCore_7_13' file was not found: '${params.schemaCore_7_13}'"
}
}
IN_schema_7_13 = Channel.fromPath(params.schemaPath_7_13)
if (params.chewbbacaJson_7_13 == true){
jsonOpt = "--json"
} else {
jsonOpt = ""
}
if (params.chewbbacaTraining_7_13){
training = "--ptf ${params.chewbbacaTraining_7_13}"
} else {
training = ""
}
// If chewbbaca is executed in batch mode, wait for all assembly files
// to be collected on the input channel, and only then execute chewbbaca
// providing all samples simultaneously
if (params.chewbbacaBatch_7_13) {
process chewbbaca_batch_7_13 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 7_13 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 7_13 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 7_13 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId chewbbaca_7_13 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
maxForks 1
scratch false
if (params.chewbbacaQueue_7_13 != null) {
queue "${params.chewbbacaQueue_7_13}"
}
publishDir "results/chewbbaca_alleleCall_7_13/", mode: "copy"
input:
file assembly from chewbbaca_in_3_8.map{ it[1] }.collect()
each file(schema) from IN_schema_7_13
output:
file 'chew_results*'
file 'cgMLST.tsv' optional true into chewbbacaProfile_7_13
set val('single'), val("7_13_chewbbaca"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_chewbbaca_7_13
set val('single'), val("chewbbaca_7_13"), val("7_13"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_chewbbaca_7_13
file ".versions"
script:
"""
{
set -x
if [ -d "$schema/temp" ];
then
rm -r $schema/temp
fi
if [ "$params.schemaSelectedLoci_7_13" = "null" ];
then
inputGenomes=$schema
else
inputGenomes=${params.schemaSelectedLoci_7_13}
fi
echo $assembly | tr " " "\n" >> input_file.txt
chewBBACA.py AlleleCall -i input_file.txt -g \$inputGenomes -o chew_results $jsonOpt --cpu $task.cpus $training
if [ "$jsonOpt" = "--json" ]; then
merge_json.py ${params.schemaCore_7_13} chew_results/*/results*
else
cp chew_results*/*/results_alleles.tsv cgMLST.tsv
fi
} || {
echo fail > .status
}
"""
}
} else {
process chewbbaca_7_13 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 7_13 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 7_13 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 7_13 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId chewbbaca_7_13 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
maxForks 1
tag { sample_id }
scratch true
if (params.chewbbacaQueue_7_13 != null) {
queue "${params.chewbbacaQueue_7_13}"
}
publishDir "results/chewbbaca_alleleCall_7_13/", mode: "copy"
input:
set sample_id, file(assembly) from chewbbaca_in_3_8
each file(schema) from IN_schema_7_13
output:
file 'chew_results*'
file '*_cgMLST.tsv' optional true into chewbbacaProfile_7_13
set sample_id, val("7_13_chewbbaca"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_chewbbaca_7_13
set sample_id, val("chewbbaca_7_13"), val("7_13"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_chewbbaca_7_13
file ".versions"
script:
"""
{
set -x
if [ -d "$schema/temp" ];
then
rm -r $schema/temp
fi
if [ "$params.schemaSelectedLoci_7_13" = "null" ];
then
inputGenomes=$schema
else
inputGenomes=${params.schemaSelectedLoci_7_13}
fi
echo $assembly >> input_file.txt
chewBBACA.py AlleleCall -i input_file.txt -g \$inputGenomes -o chew_results_${sample_id} $jsonOpt --cpu $task.cpus $training --fc
if [ "$jsonOpt" = "--json" ]; then
merge_json.py ${params.schemaCore_7_13} chew_results_*/*/results* ${sample_id}
else
mv chew_results_*/*/results_alleles.tsv ${sample_id}_cgMLST.tsv
fi
} || {
echo fail > .status
}
"""
}
}
process chewbbacaExtractMLST_7_13 {
publishDir "results/chewbbaca_7_13/", mode: "copy", overwrite: true
input:
file profiles from chewbbacaProfile_7_13.collect()
output:
file "results/cgMLST.tsv"
"""
head -n1 ${profiles[0]} > chewbbaca_profiles.tsv
awk 'FNR == 2' $profiles >> chewbbaca_profiles.tsv
chewBBACA.py ExtractCgMLST -i chewbbaca_profiles.tsv -o results -p $params.chewbbacaProfilePercentage_7_13
"""
}
if ( !params.spadesMinCoverage_8_14.toString().isNumber() ){
exit 1, "'spadesMinCoverage_8_14' parameter must be a number. Provided value: '${params.spadesMinCoverage_8_14}'"
}
if ( !params.spadesMinKmerCoverage_8_14.toString().isNumber()){
exit 1, "'spadesMinKmerCoverage_8_14' parameter must be a number. Provided value: '${params.spadesMinKmerCoverage_8_14}'"
}
IN_spades_opts_8_14 = Channel.value(
[params.spadesMinCoverage_8_14,
params.spadesMinKmerCoverage_8_14
])
if ( params.spadesKmers_8_14.toString().split(" ").size() <= 1 ){
if (params.spadesKmers_8_14.toString() != 'auto'){
exit 1, "'spadesKmers_8_14' parameter must be a sequence of space separated numbers or 'auto'. Provided value: ${params.spadesKmers_8_14}"
}
}
IN_spades_kmers_8_14 = Channel.value(params.spadesKmers_8_14)
clear = params.clearInput_8_14 ? "true" : "false"
disable_rr = params.disableRR_8_14 ? "true" : "false"
checkpointClear_8_14 = Channel.value(clear)
disableRR_8_14 = Channel.value(disable_rr)
process spades_8_14 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 8_14 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 8_14 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 8_14 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId spades_8_14 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir 'results/assembly/spades_8_14/', pattern: '*_spades*.fasta', mode: 'copy'
publishDir "results/assembly/spades_8_14/$sample_id", pattern: "*.gfa", mode: "copy"
publishDir "results/assembly/spades_8_14/$sample_id", pattern: "*.fastg", mode: "copy"
input:
set sample_id, file(fastq_pair), max_len from spades_in_5_9.join(SIDE_max_len_8_14)
val opts from IN_spades_opts_8_14
val kmers from IN_spades_kmers_8_14
val clear from checkpointClear_8_14
val disable_rr from disableRR_8_14
output:
set sample_id, file('*_spades*.fasta') into spades_out_8_9
file "*.fastg" optional true
file "*.gfa" into gfa1_8_14
set sample_id, val("8_14_spades"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_spades_8_14
set sample_id, val("spades_8_14"), val("8_14"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_spades_8_14
file ".versions"
script:
template "spades.py"
}
clear = params.clearInput_9_15 ? "true" : "false"
checkpointClear_9_15 = Channel.value(clear)
process skesa_9_15 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 9_15 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 9_15 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 9_15 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId skesa_9_15 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir 'results/assembly/skesa_9_15', pattern: '*skesa*.fasta', mode: 'copy'
input:
set sample_id, file(fastq_pair) from skesa_in_5_10
val clear from checkpointClear_9_15
output:
set sample_id, file('*.fasta') into skesa_out_9_10
set sample_id, val("9_15_skesa"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_skesa_9_15
set sample_id, val("skesa_9_15"), val("9_15"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_skesa_9_15
file ".versions"
script:
template "skesa.py"
}
if ( !params.minAssemblyCoverage_8_16.toString().isNumber() ){
if (params.minAssemblyCoverage_8_16.toString() != 'auto'){
exit 1, "'minAssemblyCoverage_8_16' parameter must be a number or 'auto'. Provided value: ${params.minAssemblyCoverage_8_16}"
}
}
if ( !params.AMaxContigs_8_16.toString().isNumber() ){
exit 1, "'AMaxContigs_8_16' parameter must be a number. Provide value: '${params.AMaxContigs_8_16}'"
}
IN_assembly_mapping_opts_8_16 = Channel.value([params.minAssemblyCoverage_8_16,params.AMaxContigs_8_16])
IN_genome_size_8_16 = Channel.value(params.genomeSize_8_16)
process assembly_mapping_8_16 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 8_16 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 8_16 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 8_16 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_8_16 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(fastq) from spades_out_8_9.join(_LAST_fastq_8_16)
output:
set sample_id, file(assembly), 'coverages.tsv', 'coverage_per_bp.tsv', 'sorted.bam', 'sorted.bam.bai' into MAIN_am_out_8_16
set sample_id, file("coverage_per_bp.tsv") optional true into SIDE_BpCoverage_8_16
set sample_id, val("8_16_assembly_mapping"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_assembly_mapping_8_16
set sample_id, val("assembly_mapping_8_16"), val("8_16"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_assembly_mapping_8_16
file ".versions"
script:
"""
{
echo [DEBUG] BUILDING BOWTIE INDEX FOR ASSEMBLY: $assembly >> .command.log 2>&1
bowtie2-build --threads ${task.cpus} $assembly genome_index >> .command.log 2>&1
echo [DEBUG] MAPPING READS FROM $fastq >> .command.log 2>&1
bowtie2 -q --very-sensitive-local --threads ${task.cpus} -x genome_index -1 ${fastq[0]} -2 ${fastq[1]} -S mapping.sam >> .command.log 2>&1
echo [DEBUG] CONVERTING AND SORTING SAM TO BAM >> .command.log 2>&1
samtools sort -o sorted.bam -O bam -@ ${task.cpus} mapping.sam && rm *.sam >> .command.log 2>&1
echo [DEBUG] CREATING BAM INDEX >> .command.log 2>&1
samtools index sorted.bam >> .command.log 2>&1
echo [DEBUG] ESTIMATING READ DEPTH >> .command.log 2>&1
parallel -j ${task.cpus} samtools depth -ar {} sorted.bam \\> {}.tab ::: \$(grep ">" $assembly | cut -c 2- | tr " " "_")
# Insert 0 coverage count in empty files. See Issue #2
echo [DEBUG] REMOVING EMPTY FILES >> .command.log 2>&1
find . -size 0 -print0 | xargs -0 -I{} sh -c 'echo -e 0"\t"0"\t"0 > "{}"'
echo [DEBUG] COMPILING COVERAGE REPORT >> .command.log 2>&1
parallel -j ${task.cpus} echo -n {.} '"\t"' '&&' cut -f3 {} '|' paste -sd+ '|' bc >> coverages.tsv ::: *.tab
cat *.tab > coverage_per_bp.tsv
rm *.tab
if [ -f "coverages.tsv" ]
then
echo pass > .status
else
echo fail > .status
fi
echo -n "" > .report.json
echo -n "" > .versions
} || {
echo fail > .status
}
"""
}
/** PROCESS_ASSEMBLY_MAPPING - MAIN
Processes the results from the assembly_mapping process and filters the
assembly contigs based on coverage and length thresholds.
*/
process process_assembly_mapping_8_16 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 8_16 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 8_16 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 8_16 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_8_16 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(assembly), file(coverage), file(coverage_bp), file(bam_file), file(bam_index) from MAIN_am_out_8_16
val opts from IN_assembly_mapping_opts_8_16
val gsize from IN_genome_size_8_16
output:
set sample_id, '*_filt.fasta', 'filtered.bam', 'filtered.bam.bai' into assembly_mapping_out_8_11_dep
set sample_id, val("8_16_process_am"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_process_am_8_16
set sample_id, val("process_am_8_16"), val("8_16"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_process_am_8_16
file ".versions"
script:
template "process_assembly_mapping.py"
}
SIDE_BpCoverage_8_16.set{ SIDE_BpCoverage_8_17 }
clear = params.clearInput_8_17 ? "true" : "false"
checkpointClear_8_17 = Channel.value(clear)
process pilon_8_17 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 8_17 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 8_17 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 8_17 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_8_17 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
echo false
publishDir 'results/assembly/pilon_8_17/', mode: 'copy', pattern: "*.fasta"
input:
set sample_id, file(assembly), file(bam_file), file(bam_index) from assembly_mapping_out_8_11_dep
val clear from checkpointClear_8_17
output:
set sample_id, '*_polished.fasta' into _pilon_out_8_11, pilon_report_8_17
set sample_id, val("8_17_pilon"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_8_17
set sample_id, val("pilon_8_17"), val("8_17"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_8_17
file ".versions"
script:
"""
{
pilon_mem=${String.valueOf(task.memory).substring(0, String.valueOf(task.memory).length() - 1).replaceAll("\\s", "")}
java -jar -Xms256m -Xmx\${pilon_mem} /NGStools/pilon-1.22.jar --genome $assembly --frags $bam_file --output ${assembly.name.replaceFirst(~/\.[^\.]+$/, '')}_polished --changes --threads $task.cpus >> .command.log 2>&1
echo pass > .status
if [ "$clear" = "true" ];
then
work_regex=".*/work/.{2}/.{30}/.*"
assembly_file=\$(readlink -f \$(pwd)/${assembly})
bam_file=\$(readlink -f \$(pwd)/${bam_file})
if [[ "\$assembly_file" =~ \$work_regex ]]; then
rm \$assembly_file \$bam_file
fi
fi
} || {
echo fail > .status
}
"""
}
process pilon_report_8_17 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
afterScript "report_POST.sh $params.projectId $params.pipelineId 8_17 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_8_17 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(coverage_bp) from pilon_report_8_17.join(SIDE_BpCoverage_8_17)
output:
file "*_assembly_report.csv" into pilon_report_out_8_17
set sample_id, val("8_17_pilon_report"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_report_8_17
set sample_id, val("pilon_report_8_17"), val("8_17"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_report_8_17
file ".versions"
script:
template "assembly_report.py"
}
process compile_pilon_report_8_17 {
publishDir "reports/assembly/pilon_8_17/", mode: 'copy'
input:
file(report) from pilon_report_out_8_17.collect()
output:
file "pilon_assembly_report.csv"
"""
echo Sample,Number of contigs,Average contig size,N50,Total assembly length,GC content,Missing data > pilon_assembly_report.csv
cat $report >> pilon_assembly_report.csv
"""
}
_pilon_out_8_11.into{ pilon_out_8_11;mlst_in_8_14;prokka_in_8_15;chewbbaca_in_8_16 }
if ( !params.minAssemblyCoverage_9_18.toString().isNumber() ){
if (params.minAssemblyCoverage_9_18.toString() != 'auto'){
exit 1, "'minAssemblyCoverage_9_18' parameter must be a number or 'auto'. Provided value: ${params.minAssemblyCoverage_9_18}"
}
}
if ( !params.AMaxContigs_9_18.toString().isNumber() ){
exit 1, "'AMaxContigs_9_18' parameter must be a number. Provide value: '${params.AMaxContigs_9_18}'"
}
IN_assembly_mapping_opts_9_18 = Channel.value([params.minAssemblyCoverage_9_18,params.AMaxContigs_9_18])
IN_genome_size_9_18 = Channel.value(params.genomeSize_9_18)
process assembly_mapping_9_18 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 9_18 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 9_18 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 9_18 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_9_18 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(fastq) from skesa_out_9_10.join(_LAST_fastq_9_18)
output:
set sample_id, file(assembly), 'coverages.tsv', 'coverage_per_bp.tsv', 'sorted.bam', 'sorted.bam.bai' into MAIN_am_out_9_18
set sample_id, file("coverage_per_bp.tsv") optional true into SIDE_BpCoverage_9_18
set sample_id, val("9_18_assembly_mapping"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_assembly_mapping_9_18
set sample_id, val("assembly_mapping_9_18"), val("9_18"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_assembly_mapping_9_18
file ".versions"
script:
"""
{
echo [DEBUG] BUILDING BOWTIE INDEX FOR ASSEMBLY: $assembly >> .command.log 2>&1
bowtie2-build --threads ${task.cpus} $assembly genome_index >> .command.log 2>&1
echo [DEBUG] MAPPING READS FROM $fastq >> .command.log 2>&1
bowtie2 -q --very-sensitive-local --threads ${task.cpus} -x genome_index -1 ${fastq[0]} -2 ${fastq[1]} -S mapping.sam >> .command.log 2>&1
echo [DEBUG] CONVERTING AND SORTING SAM TO BAM >> .command.log 2>&1
samtools sort -o sorted.bam -O bam -@ ${task.cpus} mapping.sam && rm *.sam >> .command.log 2>&1
echo [DEBUG] CREATING BAM INDEX >> .command.log 2>&1
samtools index sorted.bam >> .command.log 2>&1
echo [DEBUG] ESTIMATING READ DEPTH >> .command.log 2>&1
parallel -j ${task.cpus} samtools depth -ar {} sorted.bam \\> {}.tab ::: \$(grep ">" $assembly | cut -c 2- | tr " " "_")
# Insert 0 coverage count in empty files. See Issue #2
echo [DEBUG] REMOVING EMPTY FILES >> .command.log 2>&1
find . -size 0 -print0 | xargs -0 -I{} sh -c 'echo -e 0"\t"0"\t"0 > "{}"'
echo [DEBUG] COMPILING COVERAGE REPORT >> .command.log 2>&1
parallel -j ${task.cpus} echo -n {.} '"\t"' '&&' cut -f3 {} '|' paste -sd+ '|' bc >> coverages.tsv ::: *.tab
cat *.tab > coverage_per_bp.tsv
rm *.tab
if [ -f "coverages.tsv" ]
then
echo pass > .status
else
echo fail > .status
fi
echo -n "" > .report.json
echo -n "" > .versions
} || {
echo fail > .status
}
"""
}
/** PROCESS_ASSEMBLY_MAPPING - MAIN
Processes the results from the assembly_mapping process and filters the
assembly contigs based on coverage and length thresholds.
*/
process process_assembly_mapping_9_18 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 9_18 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 9_18 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 9_18 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId assembly_mapping_9_18 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(assembly), file(coverage), file(coverage_bp), file(bam_file), file(bam_index) from MAIN_am_out_9_18
val opts from IN_assembly_mapping_opts_9_18
val gsize from IN_genome_size_9_18
output:
set sample_id, '*_filt.fasta', 'filtered.bam', 'filtered.bam.bai' into assembly_mapping_out_9_12_dep
set sample_id, val("9_18_process_am"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_process_am_9_18
set sample_id, val("process_am_9_18"), val("9_18"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_process_am_9_18
file ".versions"
script:
template "process_assembly_mapping.py"
}
SIDE_BpCoverage_9_18.set{ SIDE_BpCoverage_9_19 }
clear = params.clearInput_9_19 ? "true" : "false"
checkpointClear_9_19 = Channel.value(clear)
process pilon_9_19 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 9_19 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 9_19 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 9_19 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_9_19 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
echo false
publishDir 'results/assembly/pilon_9_19/', mode: 'copy', pattern: "*.fasta"
input:
set sample_id, file(assembly), file(bam_file), file(bam_index) from assembly_mapping_out_9_12_dep
val clear from checkpointClear_9_19
output:
set sample_id, '*_polished.fasta' into pilon_out_9_12, pilon_report_9_19
set sample_id, val("9_19_pilon"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_9_19
set sample_id, val("pilon_9_19"), val("9_19"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_9_19
file ".versions"
script:
"""
{
pilon_mem=${String.valueOf(task.memory).substring(0, String.valueOf(task.memory).length() - 1).replaceAll("\\s", "")}
java -jar -Xms256m -Xmx\${pilon_mem} /NGStools/pilon-1.22.jar --genome $assembly --frags $bam_file --output ${assembly.name.replaceFirst(~/\.[^\.]+$/, '')}_polished --changes --threads $task.cpus >> .command.log 2>&1
echo pass > .status
if [ "$clear" = "true" ];
then
work_regex=".*/work/.{2}/.{30}/.*"
assembly_file=\$(readlink -f \$(pwd)/${assembly})
bam_file=\$(readlink -f \$(pwd)/${bam_file})
if [[ "\$assembly_file" =~ \$work_regex ]]; then
rm \$assembly_file \$bam_file
fi
fi
} || {
echo fail > .status
}
"""
}
process pilon_report_9_19 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
afterScript "report_POST.sh $params.projectId $params.pipelineId 9_19 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId pilon_9_19 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
}
tag { sample_id }
input:
set sample_id, file(assembly), file(coverage_bp) from pilon_report_9_19.join(SIDE_BpCoverage_9_19)
output:
file "*_assembly_report.csv" into pilon_report_out_9_19
set sample_id, val("9_19_pilon_report"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_pilon_report_9_19
set sample_id, val("pilon_report_9_19"), val("9_19"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_pilon_report_9_19
file ".versions"
script:
template "assembly_report.py"
}
process compile_pilon_report_9_19 {
publishDir "reports/assembly/pilon_9_19/", mode: 'copy'
input:
file(report) from pilon_report_out_9_19.collect()
output:
file "pilon_assembly_report.csv"
"""
echo Sample,Number of contigs,Average contig size,N50,Total assembly length,GC content,Missing data > pilon_assembly_report.csv
cat $report >> pilon_assembly_report.csv
"""
}
if ( params.abricateDataDir_9_20 ){
if ( !file(params.abricateDataDir_9_20).exists() ){
exit 1, "'abricateDataDir_9_20' data directory was not found: '${params.abricateDatabases_9_20}'"
}
dataDirOpt = "--datadir ${params.abricateDataDir_9_20}"
} else {
dataDirOpt = ""
}
if ( !params.abricateMinId_9_20.toString().isNumber() ){
exit 1, "'abricateMinId_9_20' parameter must be a number. Provide value: '${params.abricateMinId_9_20}'"
}
if ( !params.abricateMinCov_9_20.toString().isNumber() ){
exit 1, "'abricateMinCov_9_20' parameter must be a number. Provide value: '${params.abricateMinCov_9_20}'"
}
process abricate_9_20 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 9_20 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 9_20 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 9_20 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId abricate_9_20 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { "${sample_id} ${db}" }
publishDir "results/annotation/abricate_9_20/${sample_id}"
input:
set sample_id, file(assembly) from pilon_out_9_12
each db from params.abricateDatabases_9_20
val min_id from Channel.value(params.abricateMinId_9_20)
val min_cov from Channel.value(params.abricateMinCov_9_20)
output:
file '*.tsv' into abricate_out_9_20
set sample_id, val("9_20_abricate_$db"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_abricate_9_20
set sample_id, val("abricate_9_20_$db"), val("9_20"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_abricate_9_20
file ".versions"
script:
"""
{
# Run abricate
abricate $dataDirOpt --minid $min_id --mincov $min_cov --db $db $assembly > ${sample_id}_abr_${db}.tsv
echo pass > .status
} || {
echo fail > .status
}
"""
}
process process_abricate_9_20 {
tag "process_abricate_9_20"
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
afterScript "report_POST.sh $params.projectId $params.pipelineId 9_20 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId abricate_9_20 \"$params.platformSpecies\" false"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh"
}
input:
file abricate_file from abricate_out_9_20.collect()
output:
set val('process_abricate'), val("9_20_process_abricate"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_process_abricate_9_20
set val('process_abricate'), val("process_abricate_9_20"), val("9_20"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_process_abricate_9_20
file ".versions"
script:
template "process_abricate.py"
}
process mlst_10_21 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 10_21 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 10_21 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 10_21 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId mlst_10_21 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
// This process can only use a single CPU
cpus 1
input:
set sample_id, file(assembly) from mlst_in_8_14
output:
file '*.mlst.txt' into LOG_mlst_10_21
set sample_id, file(assembly), file(".status") into MAIN_mlst_out_10_21
set sample_id, val("10_21_mlst"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_mlst_10_21
set sample_id, val("mlst_10_21"), val("10_21"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_mlst_10_21
file ".versions"
script:
"""
{
expectedSpecies=${params.mlstSpecies_10_21}
mlst $assembly >> ${sample_id}.mlst.txt
mlstSpecies=\$(cat *.mlst.txt | cut -f2)
json_str="{'expectedSpecies':\'\$expectedSpecies\',\
'species':'\$mlstSpecies',\
'st':'\$(cat *.mlst.txt | cut -f3)',\
'tableRow':[{'sample':'${sample_id}','data':[\
{'header':'MLST species','value':'\$mlstSpecies','table':'typing'},\
{'header':'MLST ST','value':'\$(cat *.mlst.txt | cut -f3)','table':'typing'}]}]}"
echo \$json_str > .report.json
if [ ! \$mlstSpecies = \$expectedSpecies ];
then
printf fail > .status
else
printf pass > .status
fi
} || {
printf fail > .status
}
"""
}
process compile_mlst_10_21 {
publishDir "results/annotation/mlst_10_21/"
input:
file res from LOG_mlst_10_21.collect()
output:
file "mlst_report.tsv"
script:
"""
cat $res >> mlst_report.tsv
"""
}
mlst_out_10_14 = Channel.create()
MAIN_mlst_out_10_21
.filter{ it[2].text != "fail" }
.map{ [it[0], it[1]] }
.set{ mlst_out_10_14 }
IN_centre_11_22 = Channel.value(params.centre_11_22)
IN_kingdom_11_22 = Channel.value(params.kingdom_11_22)
// check if genus is provided or not
genusVar = (params.genus_11_22 == false) ? "" : "--usegenus --genus ${params.genus_11_22} "
process prokka_11_22 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 11_22 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 11_22 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 11_22 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId prokka_11_22 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
tag { sample_id }
publishDir "results/annotation/prokka_11_22/${sample_id}"
input:
set sample_id, file(assembly) from prokka_in_8_15
val centre from IN_centre_11_22
val kingdom from IN_kingdom_11_22
output:
file "${sample_id}/*"
set sample_id, val("11_22_prokka"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_prokka_11_22
set sample_id, val("prokka_11_22"), val("11_22"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_prokka_11_22
file ".versions"
script:
"""
{
prokka --outdir $sample_id --cpus $task.cpus --centre ${centre} \
--compliant --kingdom ${kingdom} ${genusVar} --increment 10 $assembly
echo pass > .status
} || {
echo fail > .status
}
"""
}
if ( !params.schemaPath_12_23 ){
exit 1, "'schemaPath_12_23' parameter missing"
}
if ( params.chewbbacaTraining_12_23){
if (!file(params.chewbbacaTraining_12_23).exists()) {
exit 1, "'chewbbacaTraining_12_23' file was not found: '${params.chewbbacaTraining_12_23}'"
}
}
if ( params.schemaSelectedLoci_12_23){
if (!file(params.schemaSelectedLoci_12_23).exists()) {
exit 1, "'schemaSelectedLoci_12_23' file was not found: '${params.schemaSelectedLoci_12_23}'"
}
}
if ( params.schemaCore_12_23){
if (!file(params.schemaCore_12_23).exists()) {
exit 1, "'schemaCore_12_23' file was not found: '${params.schemaCore_12_23}'"
}
}
IN_schema_12_23 = Channel.fromPath(params.schemaPath_12_23)
if (params.chewbbacaJson_12_23 == true){
jsonOpt = "--json"
} else {
jsonOpt = ""
}
if (params.chewbbacaTraining_12_23){
training = "--ptf ${params.chewbbacaTraining_12_23}"
} else {
training = ""
}
// If chewbbaca is executed in batch mode, wait for all assembly files
// to be collected on the input channel, and only then execute chewbbaca
// providing all samples simultaneously
if (params.chewbbacaBatch_12_23) {
process chewbbaca_batch_12_23 {
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 12_23 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 12_23 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 12_23 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId chewbbaca_12_23 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
maxForks 1
scratch false
if (params.chewbbacaQueue_12_23 != null) {
queue "${params.chewbbacaQueue_12_23}"
}
publishDir "results/chewbbaca_alleleCall_12_23/", mode: "copy"
input:
file assembly from chewbbaca_in_8_16.map{ it[1] }.collect()
each file(schema) from IN_schema_12_23
output:
file 'chew_results*'
file 'cgMLST.tsv' optional true into chewbbacaProfile_12_23
set val('single'), val("12_23_chewbbaca"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_chewbbaca_12_23
set val('single'), val("chewbbaca_12_23"), val("12_23"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_chewbbaca_12_23
file ".versions"
script:
"""
{
set -x
if [ -d "$schema/temp" ];
then
rm -r $schema/temp
fi
if [ "$params.schemaSelectedLoci_12_23" = "null" ];
then
inputGenomes=$schema
else
inputGenomes=${params.schemaSelectedLoci_12_23}
fi
echo $assembly | tr " " "\n" >> input_file.txt
chewBBACA.py AlleleCall -i input_file.txt -g \$inputGenomes -o chew_results $jsonOpt --cpu $task.cpus $training
if [ "$jsonOpt" = "--json" ]; then
merge_json.py ${params.schemaCore_12_23} chew_results/*/results*
else
cp chew_results*/*/results_alleles.tsv cgMLST.tsv
fi
} || {
echo fail > .status
}
"""
}
} else {
process chewbbaca_12_23 {
// Send POST request to platform
if ( params.platformHTTP != null ) {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH; set_dotfiles.sh; startup_POST.sh $params.projectId $params.pipelineId 12_23 $params.platformHTTP"
afterScript "final_POST.sh $params.projectId $params.pipelineId 12_23 $params.platformHTTP; report_POST.sh $params.projectId $params.pipelineId 12_23 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId chewbbaca_12_23 \"$params.platformSpecies\" true"
} else {
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; set_dotfiles.sh"
}
maxForks 1
tag { sample_id }
scratch true
if (params.chewbbacaQueue_12_23 != null) {
queue "${params.chewbbacaQueue_12_23}"
}
publishDir "results/chewbbaca_alleleCall_12_23/", mode: "copy"
input:
set sample_id, file(assembly) from chewbbaca_in_8_16
each file(schema) from IN_schema_12_23
output:
file 'chew_results*'
file '*_cgMLST.tsv' optional true into chewbbacaProfile_12_23
set sample_id, val("12_23_chewbbaca"), file(".status"), file(".warning"), file(".fail"), file(".command.log") into STATUS_chewbbaca_12_23
set sample_id, val("chewbbaca_12_23"), val("12_23"), file(".report.json"), file(".versions"), file(".command.trace") into REPORT_chewbbaca_12_23
file ".versions"
script:
"""
{
set -x
if [ -d "$schema/temp" ];
then
rm -r $schema/temp
fi
if [ "$params.schemaSelectedLoci_12_23" = "null" ];
then
inputGenomes=$schema
else
inputGenomes=${params.schemaSelectedLoci_12_23}
fi
echo $assembly >> input_file.txt
chewBBACA.py AlleleCall -i input_file.txt -g \$inputGenomes -o chew_results_${sample_id} $jsonOpt --cpu $task.cpus $training --fc
if [ "$jsonOpt" = "--json" ]; then
merge_json.py ${params.schemaCore_12_23} chew_results_*/*/results* ${sample_id}
else
mv chew_results_*/*/results_alleles.tsv ${sample_id}_cgMLST.tsv
fi
} || {
echo fail > .status
}
"""
}
}
process chewbbacaExtractMLST_12_23 {
publishDir "results/chewbbaca_12_23/", mode: "copy", overwrite: true
input:
file profiles from chewbbacaProfile_12_23.collect()
output:
file "results/cgMLST.tsv"
"""
head -n1 ${profiles[0]} > chewbbaca_profiles.tsv
awk 'FNR == 2' $profiles >> chewbbaca_profiles.tsv
chewBBACA.py ExtractCgMLST -i chewbbaca_profiles.tsv -o results -p $params.chewbbacaProfilePercentage_12_23
"""
}
/** STATUS
Reports the status of a sample in any given process.
*/
process status {
tag { sample_id }
publishDir "pipeline_status/$task_name"
input:
set sample_id, task_name, status, warning, fail, file(log) from STATUS_reads_download_1_1.mix(STATUS_integrity_coverage_2_2,STATUS_spades_2_3,STATUS_skesa_3_4,STATUS_integrity_coverage_4_5,STATUS_megahit_4_6,STATUS_megahit_fastg_4_6,STATUS_integrity_coverage_5_7,STATUS_fastqc_5_8,STATUS_fastqc_report_5_8,STATUS_trimmomatic_5_8,STATUS_assembly_mapping_3_9,STATUS_process_am_3_9,STATUS_pilon_3_10,STATUS_pilon_report_3_10,STATUS_fastqc2_5_11,STATUS_fastqc2_report_5_11,STATUS_abricate_6_12,STATUS_process_abricate_6_12,STATUS_chewbbaca_7_13,STATUS_spades_8_14,STATUS_skesa_9_15,STATUS_assembly_mapping_8_16,STATUS_process_am_8_16,STATUS_pilon_8_17,STATUS_pilon_report_8_17,STATUS_assembly_mapping_9_18,STATUS_process_am_9_18,STATUS_pilon_9_19,STATUS_pilon_report_9_19,STATUS_abricate_9_20,STATUS_process_abricate_9_20,STATUS_mlst_10_21,STATUS_prokka_11_22,STATUS_chewbbaca_12_23)
output:
file '*.status' into master_status
file '*.warning' into master_warning
file '*.fail' into master_fail
file '*.log'
"""
echo $sample_id, $task_name, \$(cat $status) > ${sample_id}_${task_name}.status
echo $sample_id, $task_name, \$(cat $warning) > ${sample_id}_${task_name}.warning
echo $sample_id, $task_name, \$(cat $fail) > ${sample_id}_${task_name}.fail
echo "\$(cat .command.log)" > ${sample_id}_${task_name}.log
"""
}
process compile_status_buffer {
input:
file status from master_status.buffer( size: 5000, remainder: true)
file warning from master_warning.buffer( size: 5000, remainder: true)
file fail from master_fail.buffer( size: 5000, remainder: true)
output:
file 'master_status_*.csv' into compile_status_buffer
file 'master_warning_*.csv' into compile_warning_buffer
file 'master_fail_*.csv' into compile_fail_buffer
"""
cat $status >> master_status_${task.index}.csv
cat $warning >> master_warning_${task.index}.csv
cat $fail >> master_fail_${task.index}.csv
"""
}
process compile_status {
publishDir 'reports/status'
input:
file status from compile_status_buffer.collect()
file warning from compile_warning_buffer.collect()
file fail from compile_fail_buffer.collect()
output:
file "*.csv"
"""
cat $status >> master_status.csv
cat $warning >> master_warning.csv
cat $fail >> master_fail.csv
"""
}
/** Reports
Compiles the reports from every process
*/
process report {
tag { sample_id }
input:
set sample_id,
task_name,
pid,
report_json,
version_json,
trace from REPORT_reads_download_1_1.mix(REPORT_integrity_coverage_2_2,REPORT_spades_2_3,REPORT_skesa_3_4,REPORT_integrity_coverage_4_5,REPORT_megahit_4_6,REPORT_megahit_fastg_4_6,REPORT_integrity_coverage_5_7,REPORT_fastqc_5_8,REPORT_fastqc_report_5_8,REPORT_trimmomatic_5_8,REPORT_assembly_mapping_3_9,REPORT_process_am_3_9,REPORT_pilon_3_10,REPORT_pilon_report_3_10,REPORT_fastqc2_5_11,REPORT_fastqc2_report_5_11,REPORT_abricate_6_12,REPORT_process_abricate_6_12,REPORT_chewbbaca_7_13,REPORT_spades_8_14,REPORT_skesa_9_15,REPORT_assembly_mapping_8_16,REPORT_process_am_8_16,REPORT_pilon_8_17,REPORT_pilon_report_8_17,REPORT_assembly_mapping_9_18,REPORT_process_am_9_18,REPORT_pilon_9_19,REPORT_pilon_report_9_19,REPORT_abricate_9_20,REPORT_process_abricate_9_20,REPORT_mlst_10_21,REPORT_prokka_11_22,REPORT_chewbbaca_12_23)
output:
file "*" optional true into master_report
"""
prepare_reports.py $report_json $version_json $trace $sample_id $task_name 1 $pid $workflow.scriptId $workflow.runName
"""
}
process compile_reports {
publishDir "pipeline_report/", mode: "copy"
if ( params.reportHTTP != null ){
beforeScript "PATH=${workflow.projectDir}/bin:\$PATH; export PATH;"
afterScript "metadata_POST.sh $params.projectId $params.pipelineId 0 $params.sampleName $params.reportHTTP $params.currentUserName $params.currentUserId 0 \"$params.platformSpecies\""
}
input:
file report from master_report.collect()
file forks from Channel.fromPath("${workflow.projectDir}/.forkTree.json")
file dag from Channel.fromPath("${workflow.projectDir}/.treeDag.json")
file js from Channel.fromPath("${workflow.projectDir}/resources/main.js.zip")
output:
file "pipeline_report.json"
file "pipeline_report.html"
file "src/main.js"
script:
template "compile_reports.py"
}
workflow.onComplete {
// Display complete message
log.info "Completed at: " + workflow.complete
log.info "Duration : " + workflow.duration
log.info "Success : " + workflow.success
log.info "Exit status : " + workflow.exitStatus
}
workflow.onError {
// Display error message
log.info "Workflow execution stopped with the following message:"
log.info " " + workflow.errorMessage
}
Cheap and disposable pipeline
Flowcraft - Easy way to create & glue lego components
flowcraft build -r denim -o my_denim_pipelineRecipes as shareable workflows created on the fly
Use cases - Innuendo
European Food Safety Authority funded project for genomic surveillance of food-borne pathogens

Use cases - Innuendo
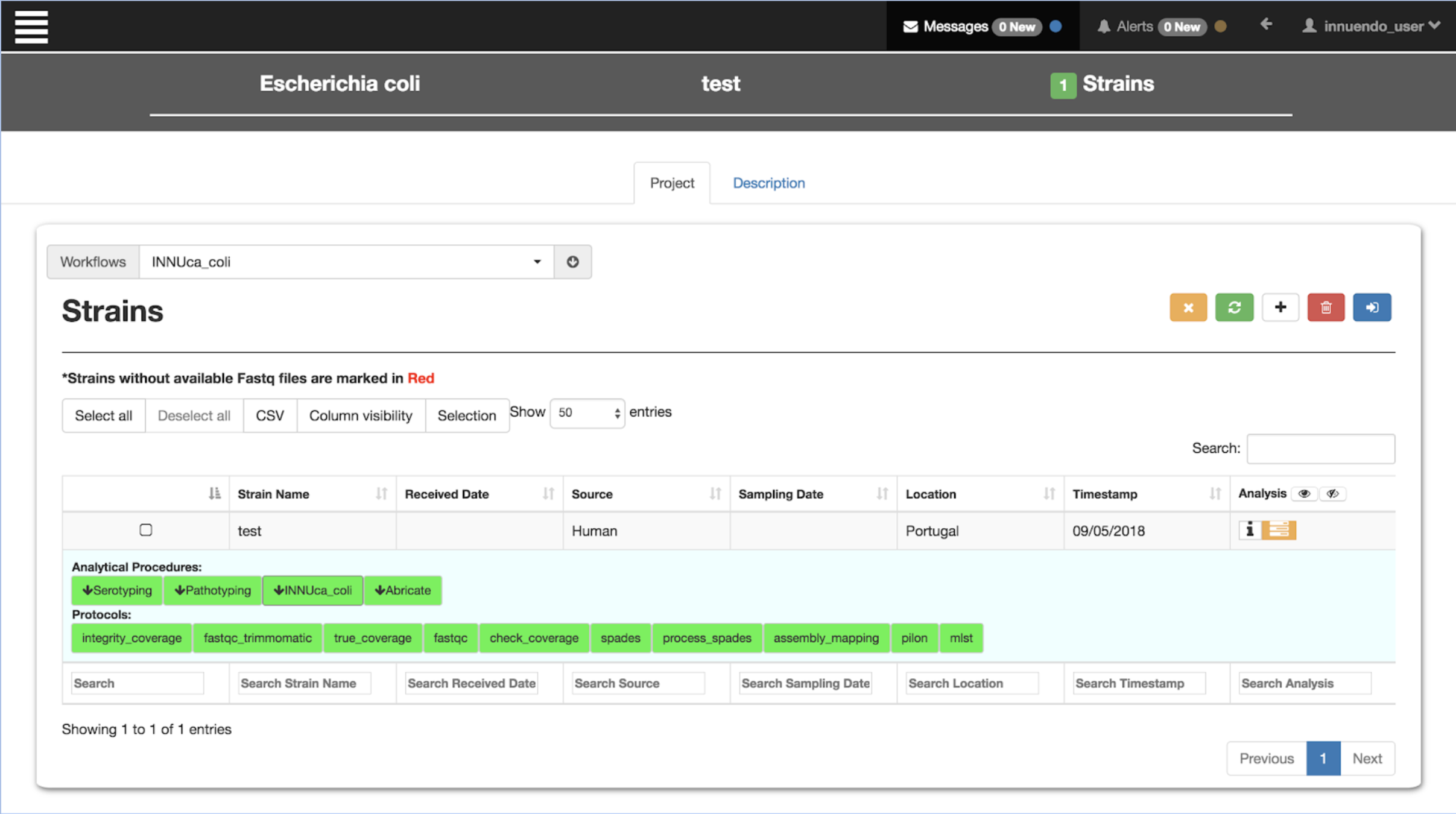
Specification of pipeline components by the user
Use cases - Genomics/epidemiology

42h on 200 CPUs
151 samples
1812 assemblies
43s/assembly


Trimming
Sampled assemblies
Use cases - Genomics/epidemiology


Dots above red line
Same sample interpreted with different profile
Potentially misinterpreted as outbreak

Wrap up
- Component-based workflow development make pipelines easy to create and mundane
- A sustainable method of creating and maintaining pipelines that is able to cope with the volatile bioinformatic software landscape
- With easy to assembly lego like components, it incentives the development of benchmarking and comparative analyses
- Much easier to contribute to the development of individual small components than massive pipelines.
Enables the creation of automatic pipelines on the fly



Diogo N Silva
Tiago F Jesus
Catarina I Mendes
Bruno
Ribeiro-Gonçalves
Core developers

Advisors
Prof. Mário Ramirez

Prof. João André Carriço
The team
Lifebit Bioinformatician
Lifebit Bioinformatician
Lifebit Bioinformatician
PhD at IMM
PI at IMM
Researcher at IMM
Thank you for your attention
and happy pipeline building
Join the fun!



conda install flowcraftpip install flowcraftBacGenTrack project [FCT / Scientific and Technological Research Council of Turkey, TUBITAK/0004/2014]
Funding and ackowledgements
ONEIDA project (LISBOA-01-0145-FEDER-016417) co-founded by 'Fundos Internacionais Europeus Estruturais e de Investimento' and the national funds from FCT - Fundação para a Ciência e Tecnologia
Lifebit Biotech Ltd.
