Interpreting evolution of SARS-CoV-2 and other viruses
Jesse Bloom
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/embo-workshop
New variants (mutants) are always arising during viral evolution




Can we predict evolutionary success of viral variants from the biochemical effects of their constituent mutations?
SARS-CoV-2's spike binds receptor and mediates viral entry
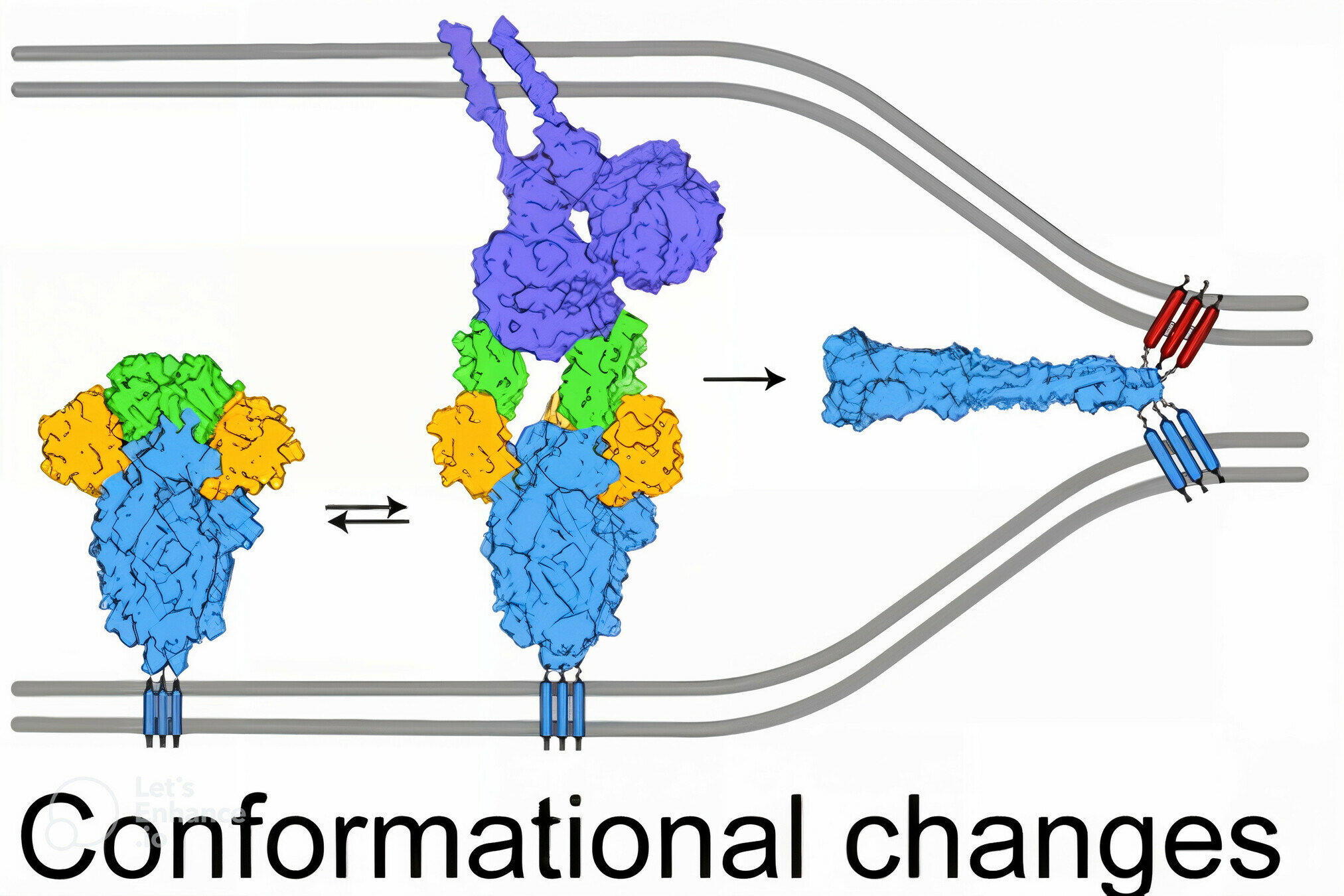
viral membrane
cell membrane
spike
spike conformational change
Image adapted from here
ACE2

antibody
Image adapted from here
SARS-CoV-2's spike binds receptor and mediates viral entry
Deep mutational scanning to measure effects of mutations

RBD
fluorescent ACE2
yeast
fluorescent tag on RBD
Yeast display of spike receptor-binding domain (RBD) to measure ACE2 binding

cell sorting
Deep mutational scanning: selections on libraries of mutants, read out by sequencing
Experiments identified N501Y as a mutation that increases ACE2 binding
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
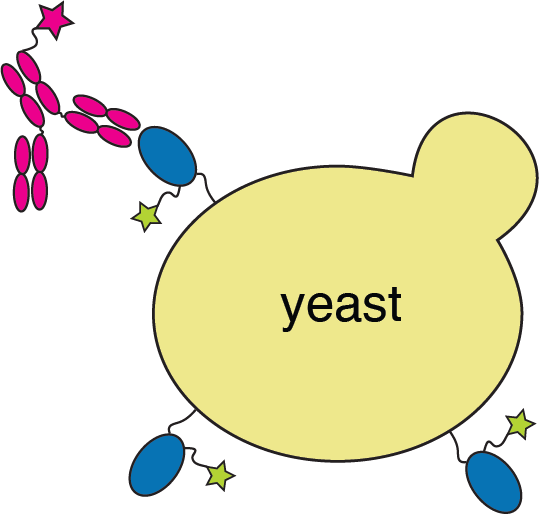
Yeast display to measure how all RBD mutations escape antibody binding

site in RBD
antibody escape
These experiments identified mutations at E484 as causing greatest antibody escape
484
These anecdotes suggest experiments can prospectively identify mutations subsequently selected by evolution
Yeast display only measures binding, which is just part of spike's biological function
cell sorting
Viral infection is more biologically relevant measure of mutation effects
However, we are cognizant of biosafety concerns of mutating actual virus

actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
Therefore we use pseudoviruses rather than actual replicative SARS-CoV-2
Pseudoviruses are modified viruses that can only undergo single round of infection

We need to link phenotype (spike mutant) and genotype (sequence encoded by virus)
To link genotype to phenotype link, we encode spike in viral genome with barcode
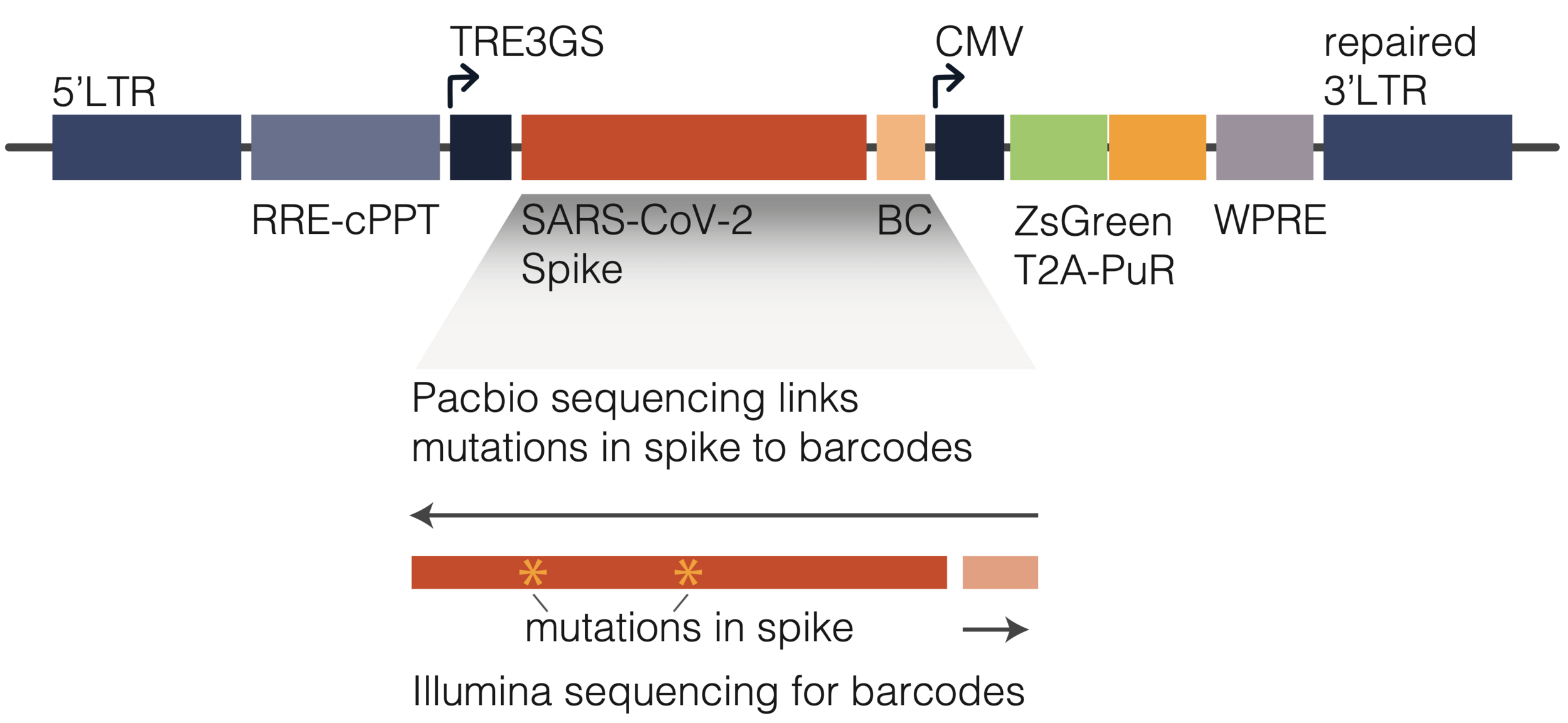
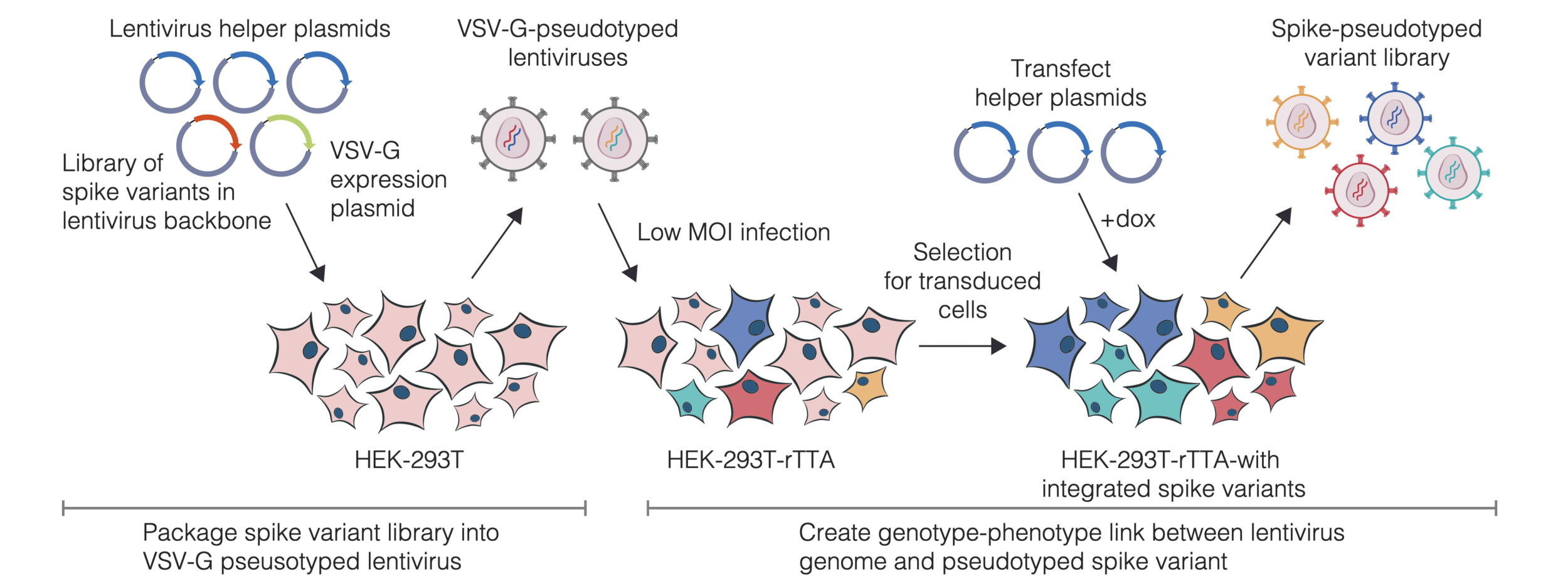
We then create genotype-phenotype linked libraries by two-step process
Result is library of spike mutant pseudoviruses with identifying barcodes
- Captures full cell-entry function of spike
- Each mutant identified by short barcode
- Pseudoviruses safe at biosafety-level 2
- Broadly applicable to viral entry proteins
Measured how spike mutations affect three molecular phenotypes
Measured how spike mutations affect three molecular phenotypes
Measured how spike mutations affect three molecular phenotypes
Neutralization by soluble ACE2 is proportional to ACE2 binding affinity
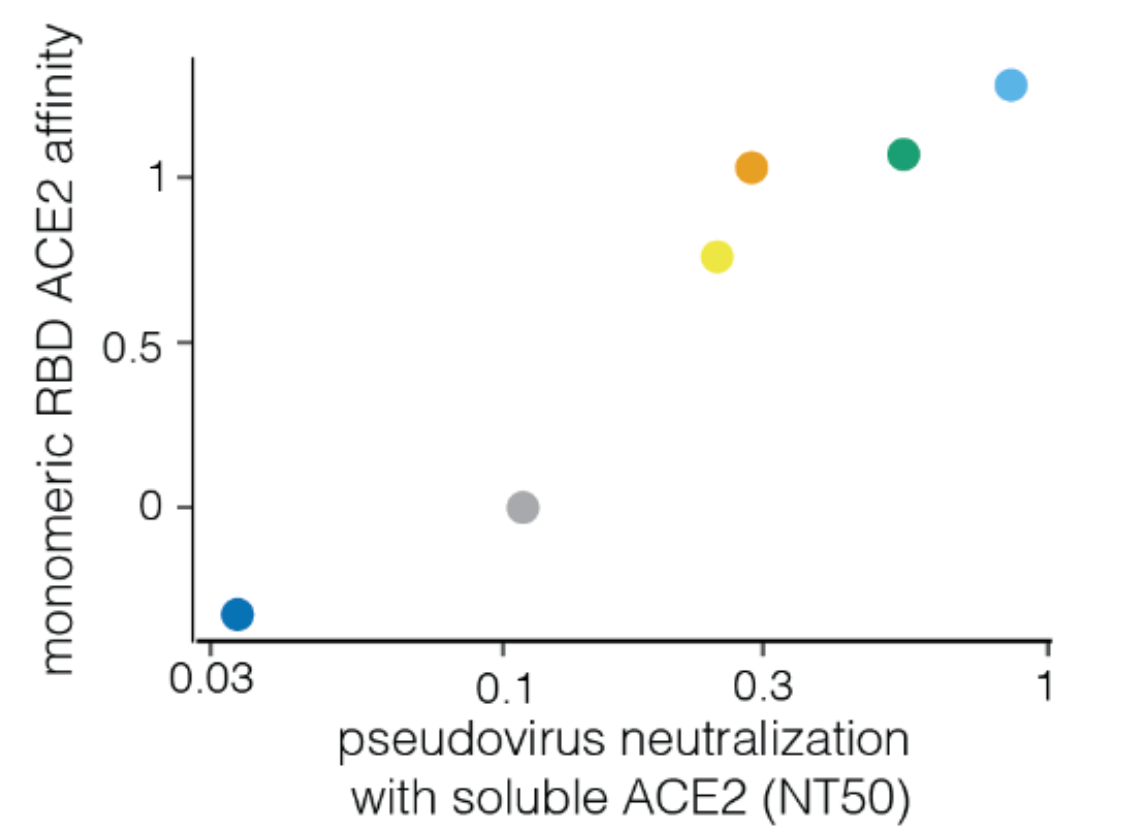
Measured how spike mutations affect three molecular phenotypes
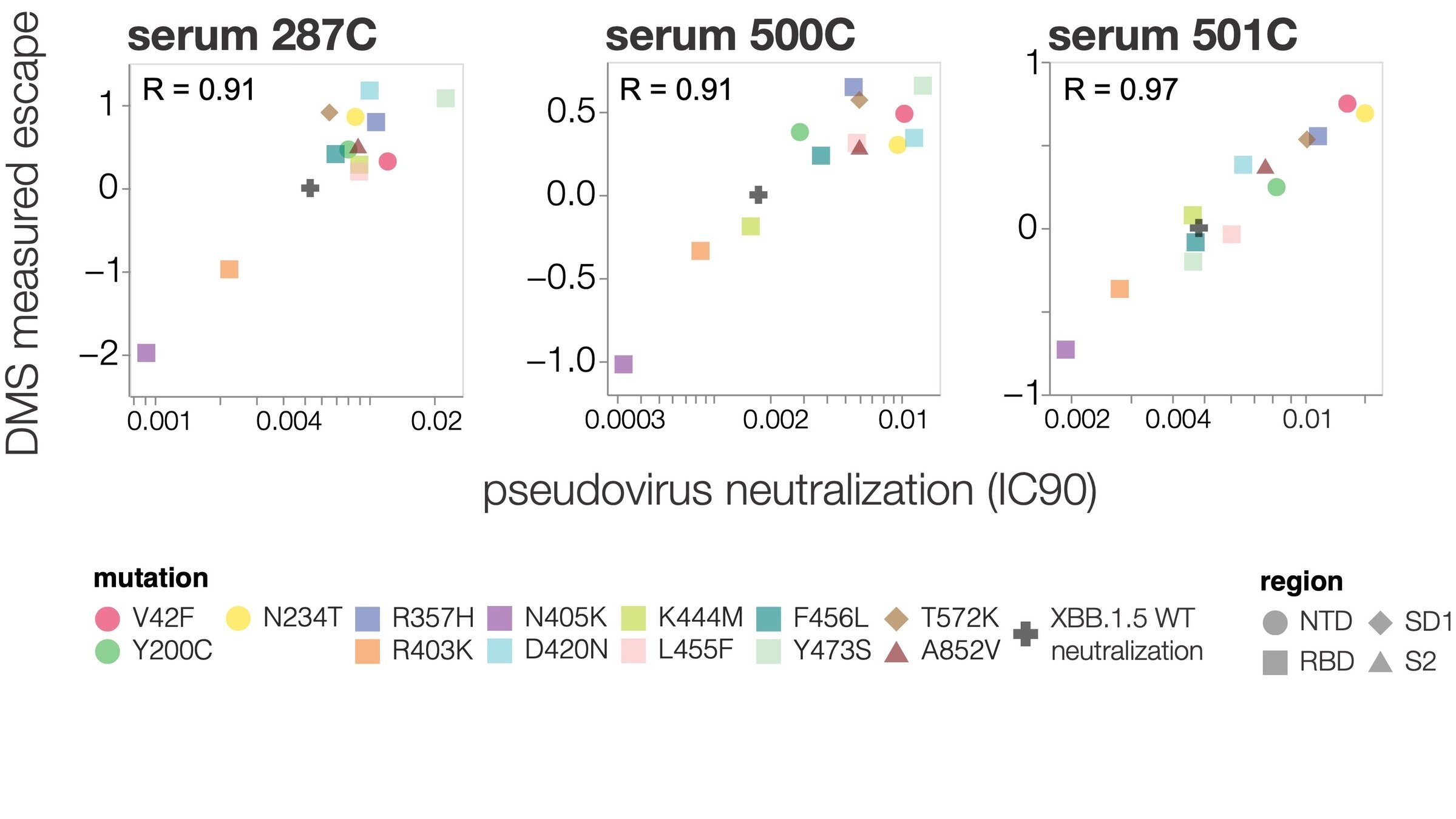
Deep mutational scanning correlates well with traditional neutralization assays
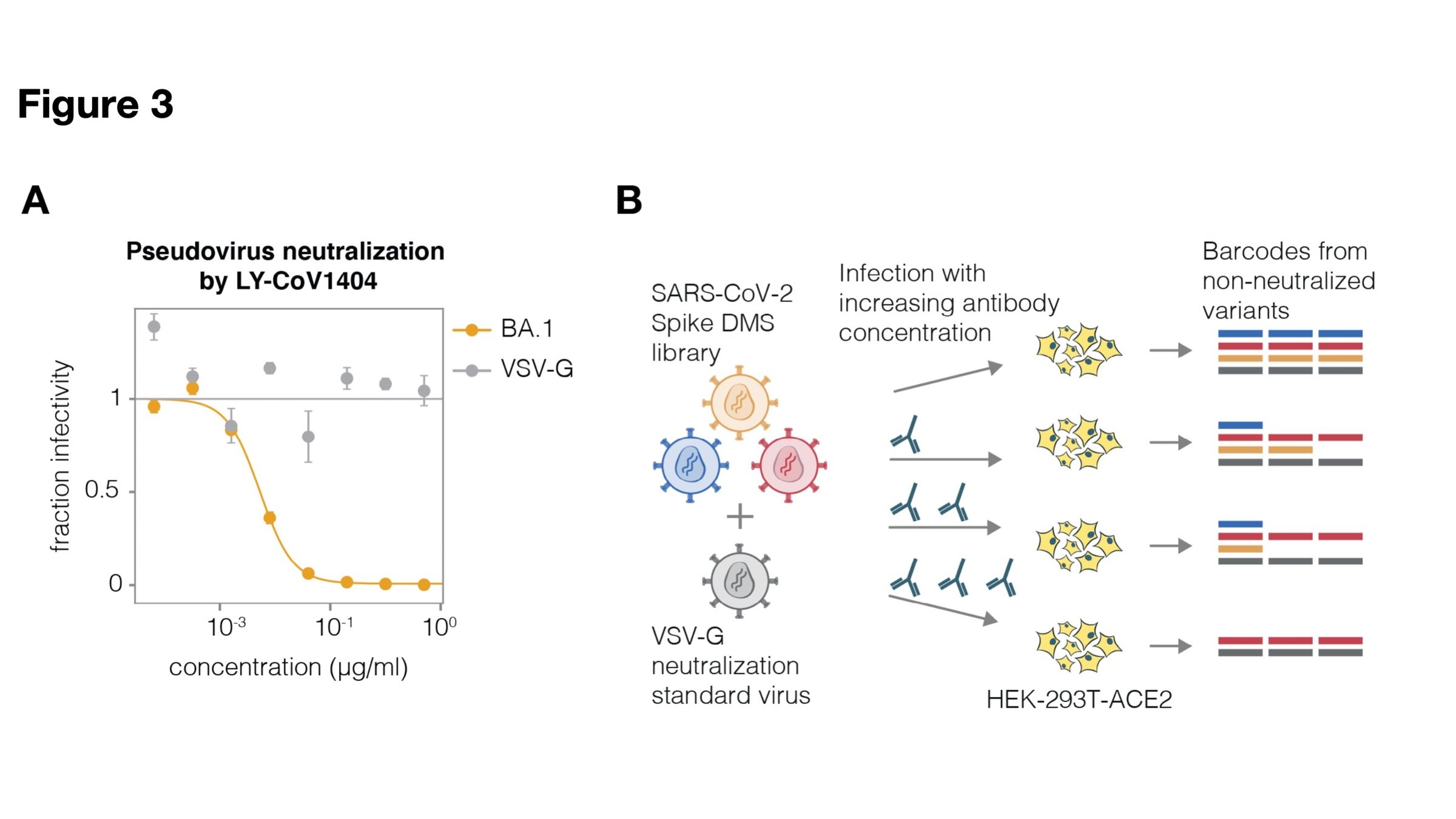
Full workflow for measure effects of mutations on antibody neutralization
How do these experimental measurements relate to actual evolution?
Estimating variant fitness (multinomial logistic regression)
With Trevor Bedford & Ben Murrell
With Trevor Bedford & Ben Murrell
Estimating variant fitness (multinomial logistic regression)
We analyze changes in clade growth rather than raw clade growth
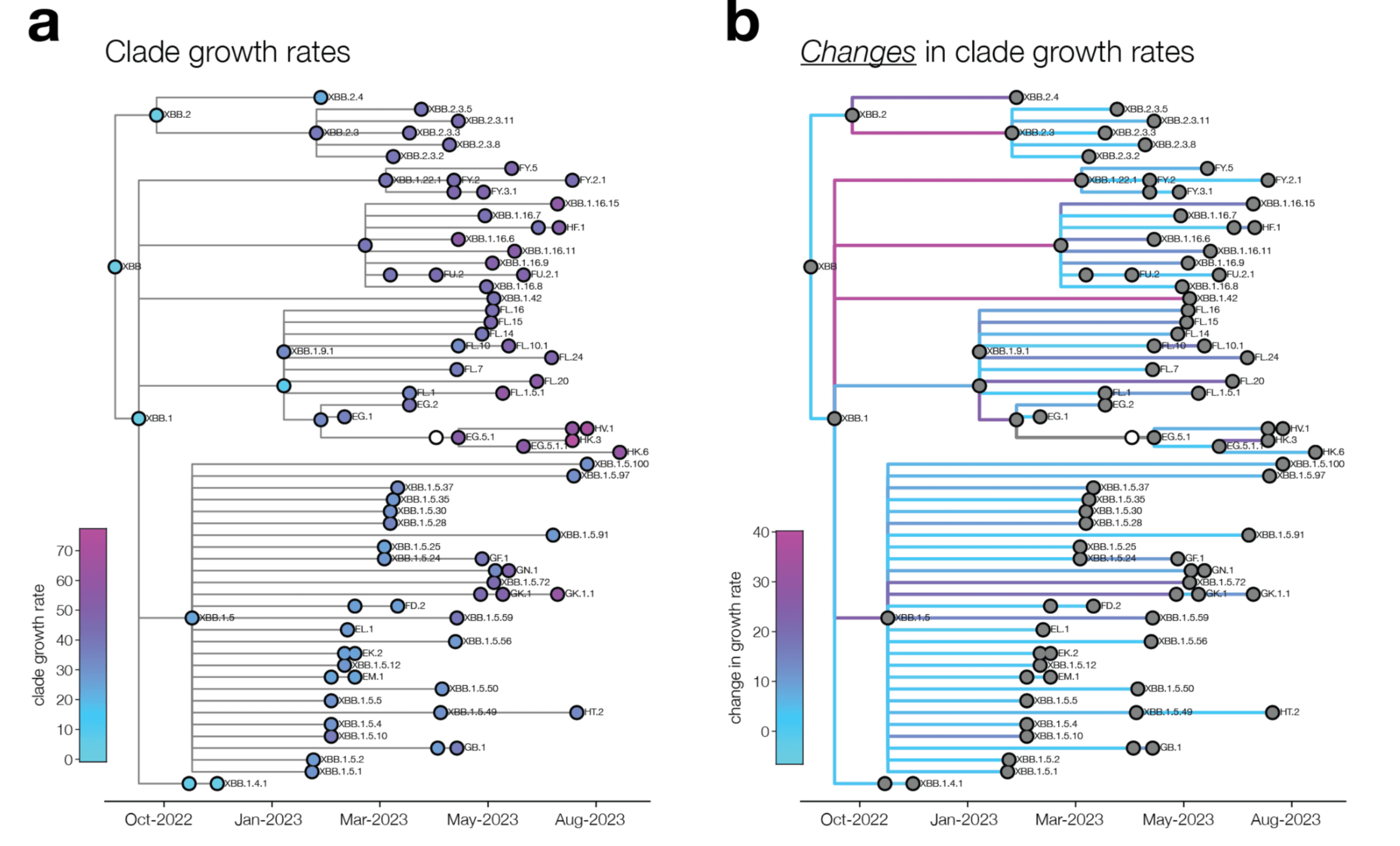
change in clade growth
clade growth
Deep mutational scanning measurements correlate with SARS-CoV-2 variant fitness
Combining phenotypes offers best predictions (because mutations can involve tradeoffs)
We can explain ~55% of the variance in growth of different clades, with largest fraction of variance uniquely explained by sera escape.
Can partially predict outcome of real-world evolutionary competition from experiments!
We can apply this experimental approach to many other viral entry proteins
- SARS-CoV-2 spike
- influenza hemagglutinin (HA)
- HIV envelope protein
- Lassa virus glycoprotein
- Nipah virus RBP and F proteins
- RSV G and F proteins
- Rabies G protein
- Chikungunya virus E proteins
Pseudovirus of clade 2.3.4.4b H5 HA
We prospectively identified A160T as strongly reducing neutralization by sera elicited by candidate vaccine strains
(L122Q, A160T, T199I)
(L122Q, P162Q, T199I)
A160T in recent human case (A160T is called A156T by CDC)
We can apply this experimental approach to many other viral entry proteins
- SARS-CoV-2 spike
- influenza hemagglutinin (HA)
- HIV envelope protein
- Lassa virus glycoprotein
- Nipah virus RBP and F proteins
- RSV G and F proteins
- Rabies G protein
- Chikungunya virus E proteins
Pseudovirus of clade 2.3.4.4b H5 HA
HA molecular phenotypes relevant to pandemic risk
HA molecular phenotypes relevant to pandemic risk
HA molecular phenotypes relevant to pandemic risk
HA molecular phenotypes relevant to pandemic risk
HA molecular phenotypes relevant to pandemic risk
These data can inform surveillance of H5 influenza viruses spreading in animals
We prospectively identified A160T as strongly reducing neutralization by sera elicited by candidate vaccine strains
(L122Q, A160T, T199I)
(L122Q, P162Q, T199I)
A160T in recent human case in Missouri
Conclusions
For human endemic (SARS-CoV-2) and potential pandemic (H5N1) viruses, we can safely measure how mutations to entry proteins affect key molecular phenotypes.
For SARS-CoV-2, these measurements can help predict success of variants in humans.
For H5N1, these measurements can help inform surveillance of viral evolution.
Bloom lab
Bernadeta Dadonaite
Kate Crawford
Caelan Radford
Tyler Starr
Allie Greaney
Rachel Eguia
William Hannon
Jenny Ahn
Fred Hutch Cancer Center
Trevor Bedford
John Huddleston
University of Washington
Helen Chu and HAARVI cohort
Neil King
David Veesler

Thanks


Pirbright Institute
Thomas Peacock
University of Pennsylvania
Scott Hensley
Louise Moncla
Jordan Ort
St Jude Children's Hospital
Richard Webby