JavaScript and Bioinformatics
- Introduction to JavaScript (ES5 + ES6)
- Node, npm ecosystem, Electron
- BioNode, BioJS and others
- Data version control with dat
- Data pipelines with gasket
- Data visualization with Plotly (D3+WebGL)
- Emscripten, asm.js
Why? What? How?
JavaScript is...
Prototype based: Functional and OO
Classes?
JavaScript classes are introduced in ECMAScript 6 and are syntactical sugar over JavaScript's existing prototype-based inheritance. The class syntax is not introducing a new object-oriented inheritance model to JavaScript. JavaScript classes provide a much simpler and clearer syntax to create objects and deal with inheritance.
- Event driven
- Non-blocking I/O
- Lightweight and Efficent
- npm package system

Events
Non-Blocking I/O
Modules
See: Art of Node



real time replication and versioning for data sets
Represent biological data on the web
modular and universal bioinformatics
bionode philosophy
- follow Unix philosophy
- be small, simple, and do one thing well
- CommonJS modules, npm
- provide callbacks, events, Node.js streams
- using streams avoids storing intermediate data on disk or running out of memory
- pipeable CLI
- client-side when possible (some modules, e.g. bionode-bwa use C/C++ libs)
A. retrieve data from web-resources
B. deal with data formats
C. handle sequences
D. wrappers (e.g. sam, bwa)module for working with NCBI APi (e-utils)
bionode-ncbi [command] [arguments] --limit (-l) --throughput (-t)Commands:
search <database> [searchTerm]
// => stream of objects found
link <sourceDatabase> <destinationDatabase>
// => stream with unique IDs linked to the passed source db unique ID
ncbi.search('genome', 'arthropoda').pipe(ncbi.expand('tax')).pipe(ncbi.plink('tax', 'sra')
// plink: take the srcID from a property of the Streamed object and attached the result to a
// property with the name of the destination DB
download <database> [searchTerm]
// currently only supports sra and assembly, accepts keyword gff for annotations
urls <database> [searchTerm]
// => {url, uid(NCBI)}. only sra, assemlby. accepts gff
expand <property> [destinationProperty]
// looks for a field named property+id (biosampleid) in the Streamed object. Then it will do a
// ncbi.search for that id and save the result under Streamed object.property.
fetch <database> <searchterm> [optional advanced params]
// => returns the records from the database that match the search term
Url for Achromyrmex Assembly
examples from sanger14
import xml.etree.ElementTree as ET
from Bio import Entrez
Entrez.email = "mail@bmpvieira.com"
esearch_handle = Entrez.esearch(db="assembly", term="Achromyrmex")
esearch_record = Entrez.read(esearch_handle)
for id in esearch_record['IdList']:
esummary_handle = Entrez.esummary(db="assembly", id=id)
esummary_record = Entrez.read(esummary_handle)
documentSummarySet = esummary_record['DocumentSummarySet']
document = documentSummarySet['DocumentSummary'][0]
metadata_XML = document['Meta'].encode('utf-8')
metadata = ET.fromstring('' + metadata_XML + '')
for entry in Metadata[1]:
print entry.textpython
javascript
var bio = require('bionode')
bio.ncbi.urls('assembly', 'Acromyrmex', function(urls) {
console.log(urls[0].genomic.fna)
})bionode-fasta --path input.fasta output.jsonin development!
check type, reverse, reverse complement, transcribe base, get codon AA, remove introns,
transcribe, translate, reverse exons, find non-canonical splice sites, check canonical
translation start site, get reading frames, get open reading frames, get all open reading
frames, find longest open reading frame
bionode with ES6/7
full list of bionode modules
"Nucleotide Bitwise Exhaustive Alignment Mapping"
1000 -> A, 0100 -> T, 0010 -> G, 0001 -> C
W = (A | T)
N = (A | T | G | C)
“ATGCATGW & ATATWWNN” =>
1000 0100 0010 0001 1000 0100 0010 1100 &
1000 0100 1000 0100 1100 1100 1111 1111
=======================================
1000 0100 0000 0000 1000 0100 0010 1100 // matches
matches |= matches >>> 1
matches |= matches >>> 2 // now rightmost of every 4 important
matches &= 0x11111111
1111 0111 1000 0000 1111 0111 1011 1111 (matches) &
0001 0001 0001 0001 0001 0001 0001 0001
=======================================
0001 0001 0000 0000 0001 0001 0001 0001
matches |= matches >>> 3;
matches |= matches >>> 6;
matches = ((matches >>> 12) & 0xF0) | (matches & 0xF)
0000 0000 0000 0000 0000 0000 1100 1111Nt.seq()
.read( [String sequenceData] ), readFASTA( [String fastaData] ),
.size(), .sequence(),
.complement(), .equivalent( [Nt.Seq compareSequence] ),
.replicate( [optional Integer offset], [optional Integer length] ),
.polymerize( [Nt.Seq sequence] ),
.insertion( [Nt.Seq insertedSequence], [Integer offset] ),
.deletion( [Nt.Seq offset], [Integer length] ),
.repeat( [Integer count] ), .mask( [Nt.Seq sequence] ),
.cover( [Nt.Seq sequence] ),
.content(), .fractionalContent(), .contentATGC(), .fractionalContentATGC(),
.translate( [optional Integer offset], [optional Integer length] ),
.translateFrame([optional Integer frame],[optional Integer AAoffset],[optional Integer AAlength]),
.mapSequence( [Nt.Seq querySequence] )
// Node only
.loadFASTA( [String pathname] ),
.load4bnt( [String pathname] ),
.save4bnt( [optional String name], [optional String path] )
Nt.Matchmap( [Nt.Seq querySeq], [Nt.Seq searchSeq] )
.results( [optional Integer offset], [optional Integer count] )
.best(), .top( [Integer count] ), .bottom( [Integer count] ), .matchFrequencyData()
Nt.MatchResult
.alignment(), .alignmentMask(), .alignmentCover(),
"Represent biological data on the web"
- maintains a registry of packages
- everything on npm
- follows conventions, e.g. docs, tests, Travis, style
- attempts to maintain a defined "common structure"
- use bio-js-events
- ultimately, any package with "biojs" keyword will become included in the "registry"
- IMO, "conventions" should be broad - as long as there is good documentation and tests, all is well
- seen by wide variety of packages/authors? - that's ok, frontend tends to be "messier" than backend
- (aside from "core biojs team" packages)
biojs: msa
import fasta, clustal; export to fASTAb; generate consensus sequence
biojs: pv

Rego, N, Koes, D. 3Dmol.js: molecular visualization with WebGL. 2014. Bioinformatics 31(8), 2015, 1322-1324
biojs: drawrnajs
biojs: cytoscape.js
biojs: biojs-muts-needleplot
biojs: biojs-vis-sequence
biojs3 draft: web components
<biojs-msa>
<biojs-io-fasta url="./Q7T2N8.fasta" />
</biojs-msa>Web components usher in a new era of web development based on encapsulated and interoperable custom elements that extend HTML itself
Mayya Sedova, Lukasz Jaroszewski, and Adam Godzik
Protael: protein data visualization library for the web.
Bioinformatics (2015) doi:10.1093/bioinformatics/btv605
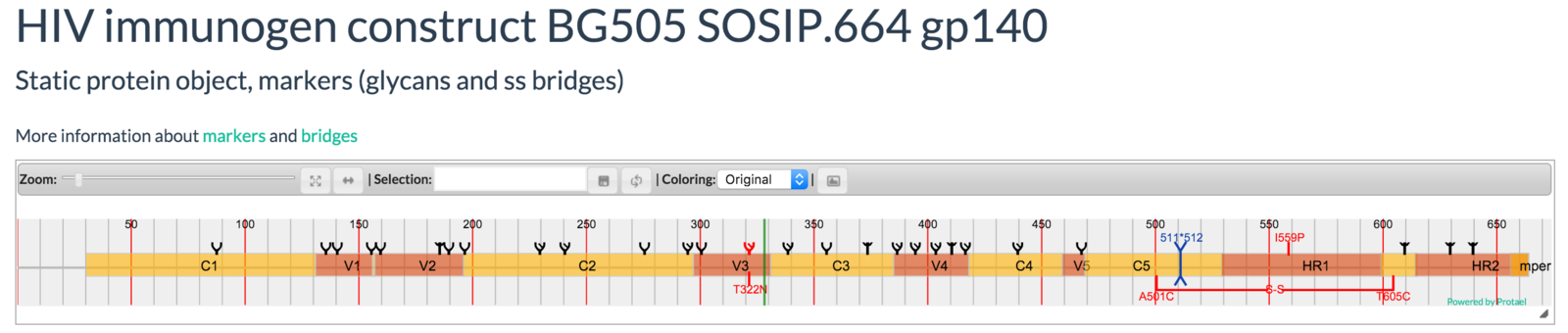
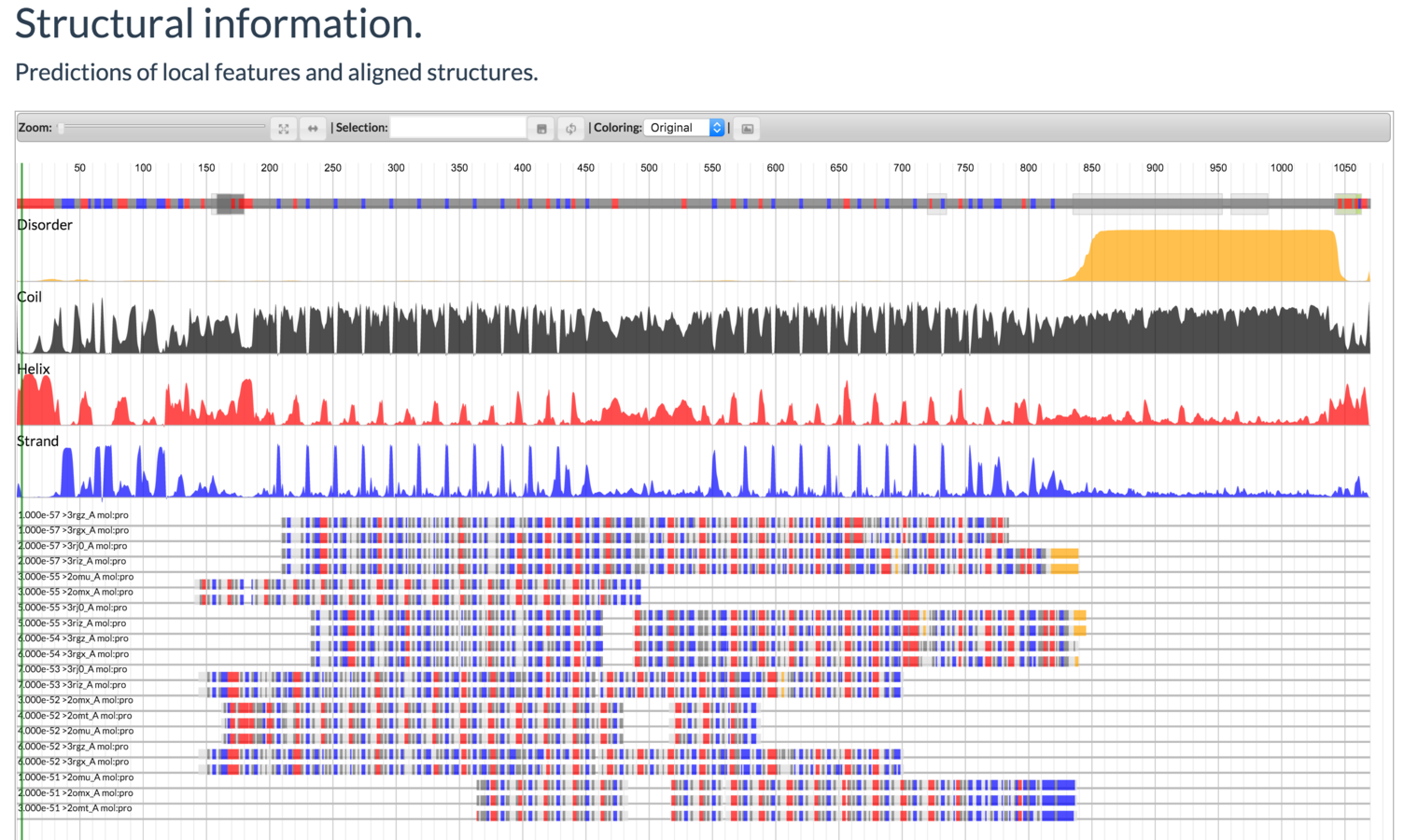
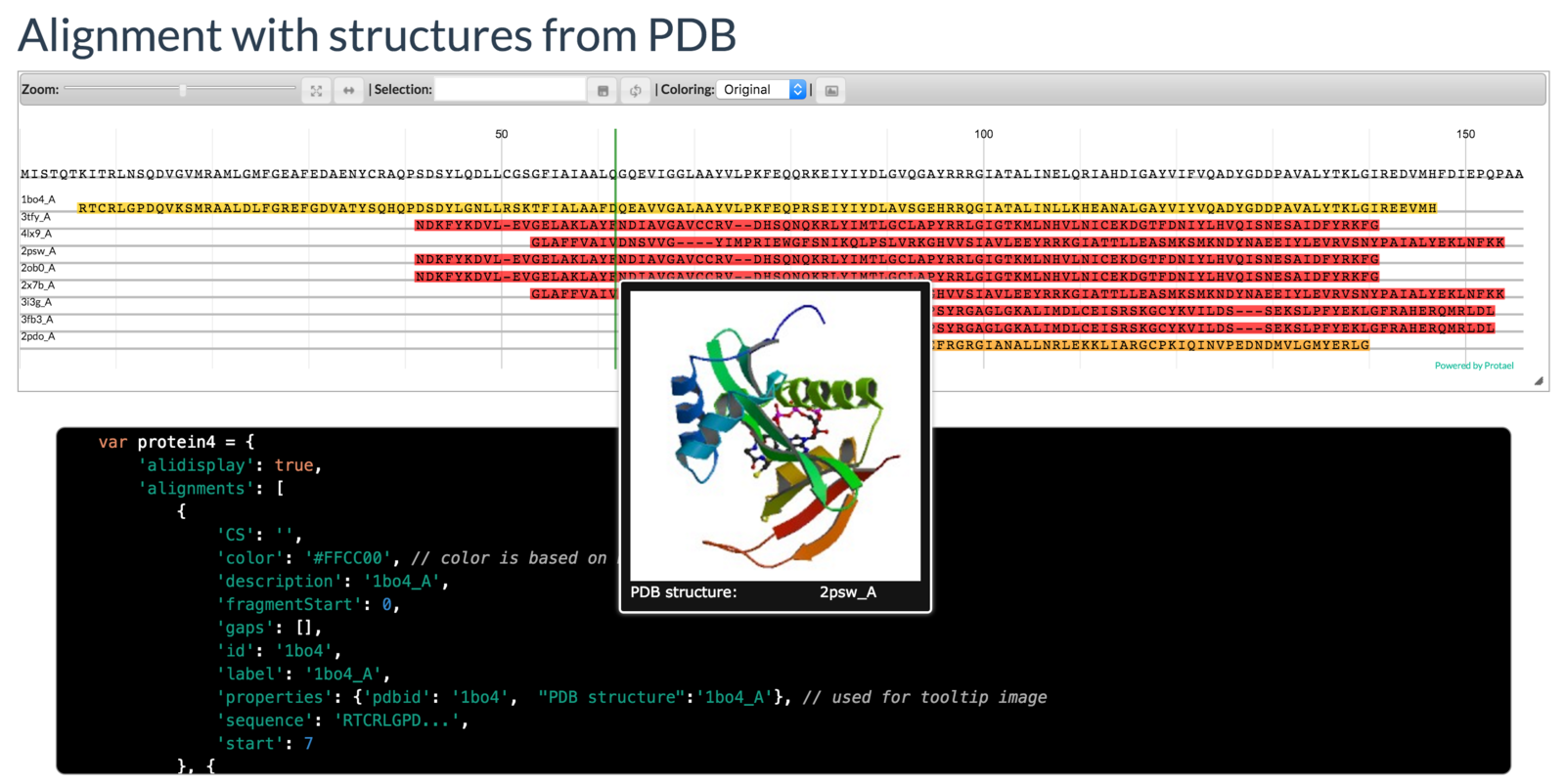
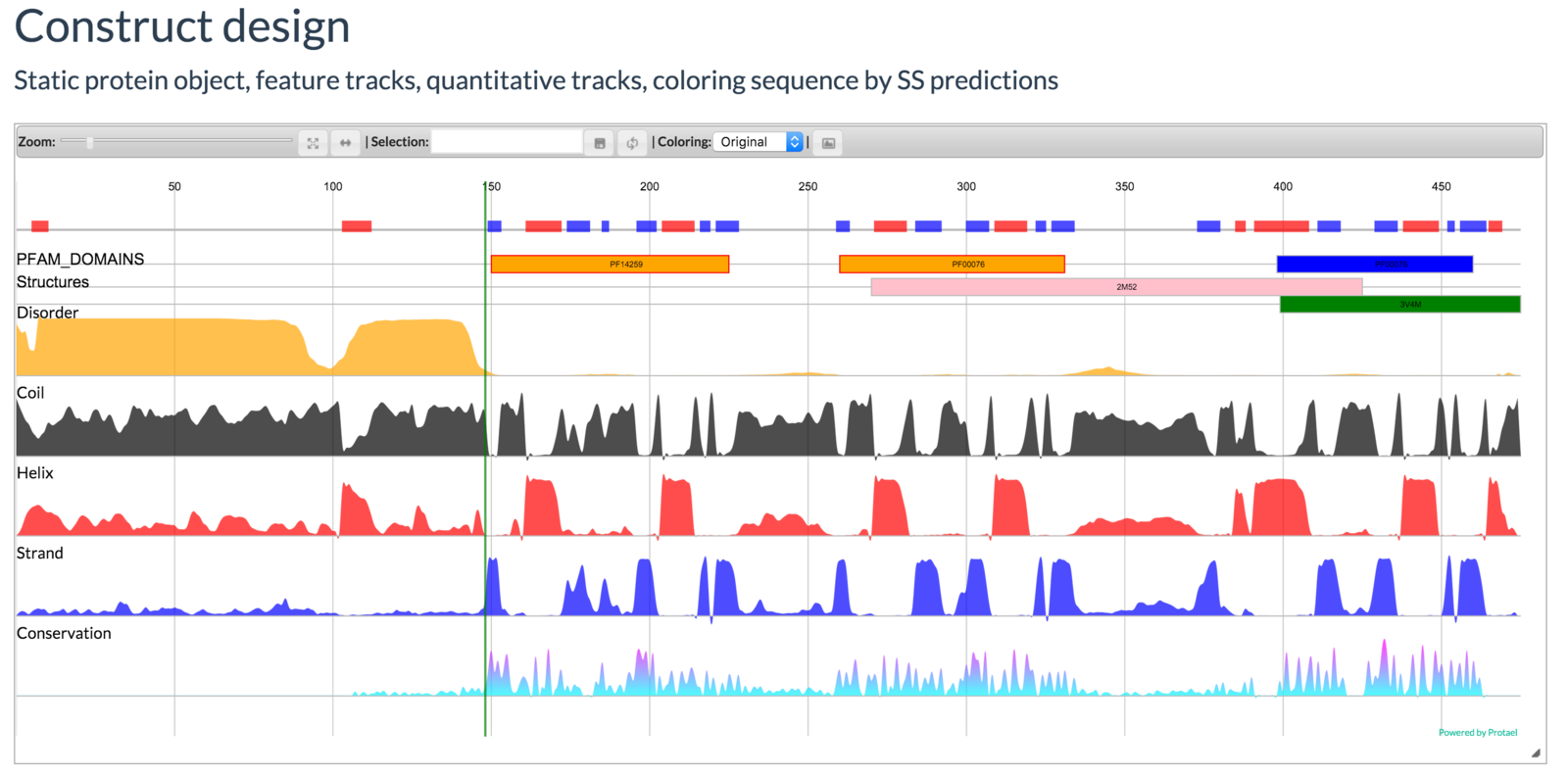
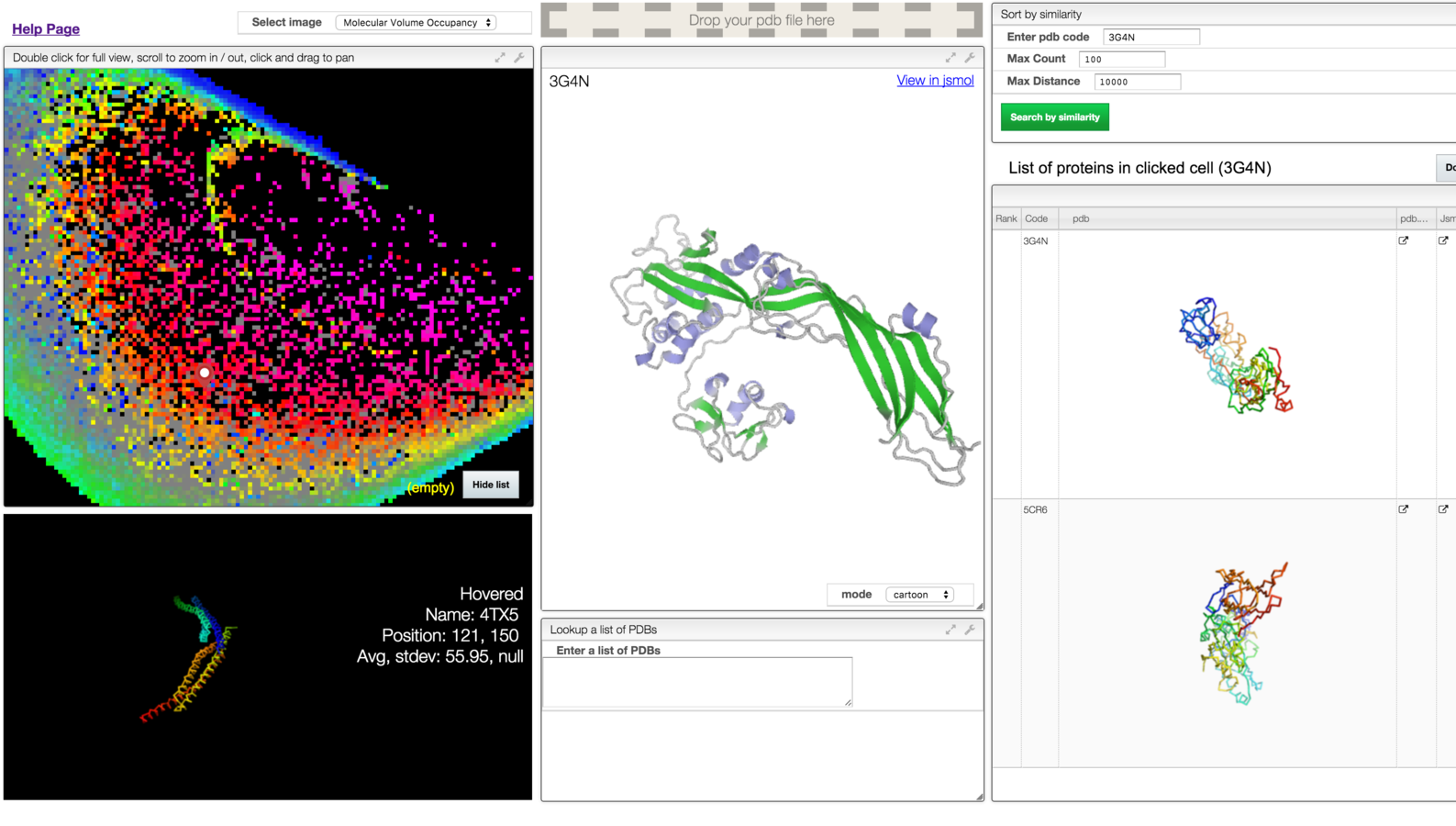
a web based interactive map of the protein data bank in shape space
dat is a version controlled, decentralized data sync tool for collaboration between data people and data systems
dat init
# takes newline delimited JSON, CSV, protocol buffers
echo '{"name": "alice", "age": "35"}' > people.json
echo '{"name":"bob", "age": "34"}' >> people.json
dat import people.json -d people --key name
# Make some changes...
dat import people.json -d people -k name
dat export -d people
# Can also write blobs (Binary Large OBjects)
dat write http://try-dat.com/static/img/wildcat.jpg -m "Added a wildcat because I can."
dat read wildcat.jpg > wildcat-is-out-of-dat.jpg
dat status
dat log
dat datasets
dat checkout <version>alternative approach to forks -> pull forks from peers
gasket.json:
{
"import-data": [
"bionode-ncbi search genome eukaryota",
"dat import -d eukaryota --key=uid"
],
"search-ncbi": [
"dat export -d eukaryota",
"grep Guillardia",
"tool-stream extractProperty assemblyid",
"bionode-ncbi download assembly -",
"tool-stream collectMatch status completed",
"tool-stream extractProperty uid",
"bionode-ncbi link assembly bioproject -",
"tool-stream extractProperty destUID",
"bionode-ncbi link bioproject sra -",
"tool-stream extractProperty destUID",
"grep 35526",
"bionode-ncbi download sra -",
"tool-stream collectMatch status completed",
"tee > metadata.json"
],
"index-and-align": [
"cat metadata.json",
"bionode-sra fastq-dump -",
"tool-stream extractProperty destFile",
"bionode-bwa mem **/*fna.gz"
],
"convert-to-bam": [
"bionode-sam 35526/SRR070675.sam"
]
}$ npm install -g gasket
$ gasket ls
import-data
search-ncbi
index-and-align
convert-to-bam
$ gasket run import-data
# ...reproducible, cross-platform data pipelines
open sourced Nov. 17 2015! announcement
takes the JSON specification of a chart and produces it as an interactive visualization
<JS, R, Python, MATLAB>
JS is slow,
so is your algorithm.
asm.js , a strict subset of JavaScript that can be used as a low-level, efficient target language for compilers

Emscripten is an LLVM-based project that compiles C and C++ into highly-optimizable JavaScript in asm.js format. This lets you run C and C++ on the web at near-native speed, without plugins.
the takeaway
- Browsers are here to stay. JS is too. JS is continually evolving. (yearly spec from ES6 onwards)
- Google is working hard to make V8 faster and faster
- JS+SVG/WebGL provides the most high quality interactive visualizations today. (and WebGL2 is around the corner). Use plotly for R,Python, MATLAB.
- npm is the world's largest collection of open source packages. (started 2009)
- emscripten/asm.js will continue to improve. (C/C++ -> JS at 1.x speed!)
- bionode/dat/gasket can create reproducible analysis pipelines
- "bionode-bwa" is the same performance as "bwa" - its a wrapper
- BioJS seems to be a mish-mash of packages who tagged it - that's ok
- Non-BioJS wep apps are great - but how to find them?
- Everyone should publish to npm with "biojs" tag && BioJS "core" team (and contribs) can continue to publish "official biojs-*" modules
- bionode provides a unified way to develop pipelines. but does not compare to bioconductor.
- thats ok! Wrap R scripts in pipeable modules!
- Node/JS is the best for dealing with web data and streams. request >>> httr
- Node has a huge open source community. Full-stack with JS for scalable web apps is the way to go. Write your RESTful API with Node, not django/flask.
- streams are well suited for data pipelines. small chunks at one time. algorithms which require all data in memory at once won't work with streams. heavy nGB data analysis? It's probably in C already. Make your own bionode wrapper! Contribute to reproducible research!
the takeaway 2
JS and the best visualizations/apps

browser, server-side, cross-platform
JS and scalable RESTful web services
world's most active packages
JS and approaching native speed
emscripten/asm.js/webassembly
bionode and tools for creating reproducible data pipelines
gasket/dat
bionode and huge repo of mature tools

bioconductor, data science python packages the clear alternatives
bionode and wrapping mature tools into streams
sra, bwa, sam, bbi in dev. atm
bionode and the potential to unify bioinformatics across all languages through the language of the web.
exercise
develop your own pipeline!
- Data acquisition with bionode-ncbi
- Data analysis with....
- bionode, BioJS, NtSeq, ...
- Python, R
- Data visualization with...
- plotly, BioJS, 3DMol, ...
try to use gasket and dat as well!
thanks!
