for beginners

Agenda:
- What is the challenge
- What is Nextflow (NF)
- Nextflow basics
- Superpowers
- Existing pipelines and good boilerplates
- Q&A
What is the challenge?
Data
Output
Data

Function
Software
- Version
- Options
Data
Function
Software
- Version
- Options
Output
Data
Any change in:
- Data
- Software version
- Command options
Can result in different output data
Results should be reproducible!
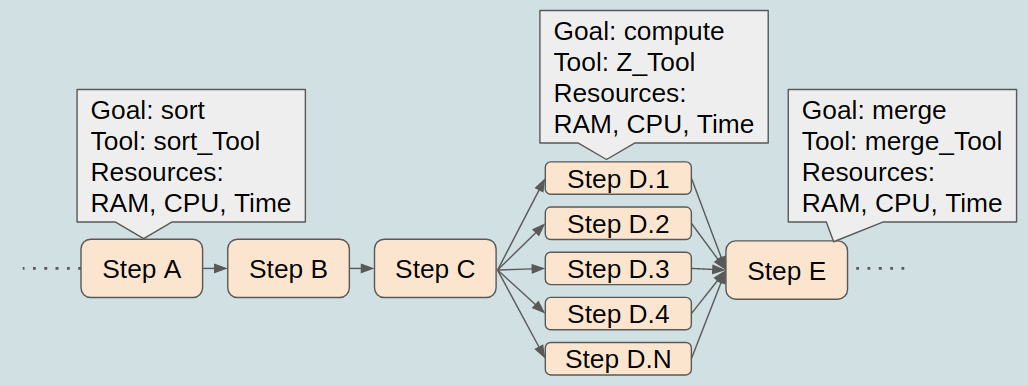

Parallelisation of tasks
is efficient

Parallelisation of tasks
is efficient

There are different HPC scheduling systems/executors
We should isolate software dependencies to give exact version of software to user
It is hard and error-prone to install software manually
Goal

Data

Packaged software

Guidelines how to process data with given software
Output
DSL - Domain specific Language
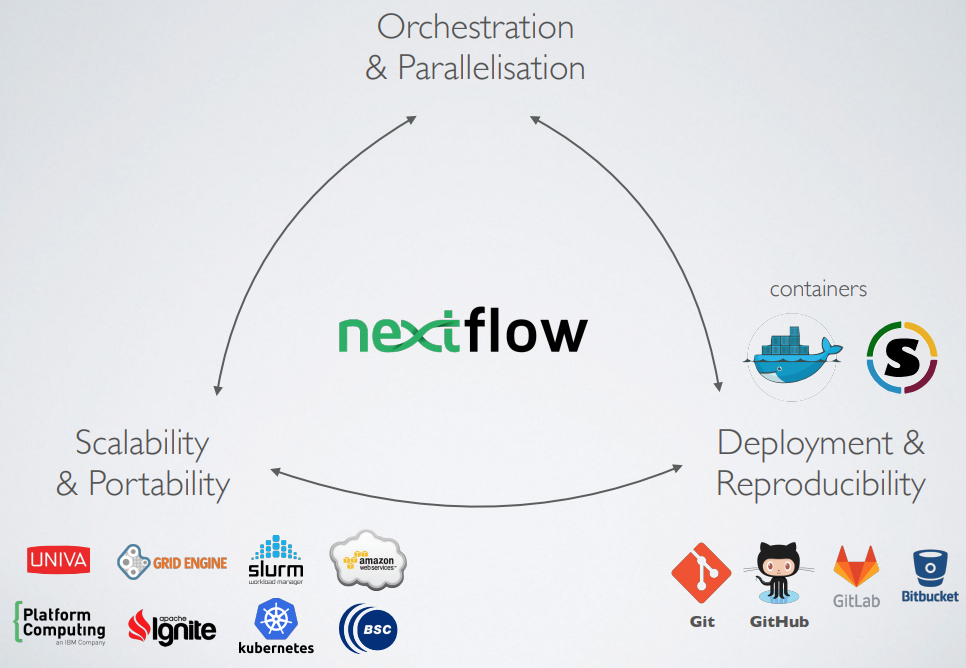
What is
Data
Software package
Guidelines how to process data with given software
Guidelines how to process data with given software using specific executor
Submit tasks in parallel to the specified executor
Output
Nextflow basics
- Processes
- Channels
Process
process makeSTARindex {
input:
file fasta from ch_fasta_for_star_index
output:
file "star" into star_index
script:
"""
mkdir star
STAR \\
--runMode genomeGenerate \\
--runThreadN ${task.cpus} \\
--sjdbGTFfile $gtf \\
--genomeDir star/ \\
--genomeFastaFiles $fasta \\
$avail_mem
"""
}- Takes input
- Executes script
- Passes output of script
- Has its own needs
- Has one specific job
- Fires automatically when input is available
Channel
process makeSTARindex {
input:
file fasta from ch_fasta_for_star_index # I am a channel
output:
file "star" into star_index # I am also a channel
script:
"""
mkdir star
STAR \\
--runMode genomeGenerate \\
--runThreadN ${task.cpus} \\
--sjdbGTFfile $gtf \\
--genomeDir star/ \\
--genomeFastaFiles $fasta \\
$avail_mem
"""
}- Passes data between processes asynchronously
- There are multiple operation you can do with channels
- merge, mix, fork, combine, divide etc.
- There are different types of channels
Channel
process plink_to_vcf{
input:
set file(bed), file(bim), file(fam) from harmonised_genotypes
output:
file "harmonised.vcf.gz" into harmonised_vcf_ch
script:
"""
plink2 --bfile ${bed.simpleName} --recode vcf-iid --out ${bed.simpleName}
bgzip harmonised.vcf
"""
}
process vcf_fixref{
input:
file input_vcf from harmonised_vcf_ch
output:
file "fixref.vcf.gz" into filter_vcf_input
script:
"""
bcftools index ${input_vcf}
bcftools +fixref ${input_vcf} -- -f ${fasta} -i ${vcf_file} | \
bcftools norm --check-ref x -f ${fasta} -Oz -o fixref.vcf.gz
"""
}Superpowers?

