OPTIMISING TRANSCRIPTOME ASSEMBLY
Richard Smith-Unna
![]()
MAYBE YOU DON'T NEED
TRANSCRIPTOME ASSEMBLY
- transcriptome assembly is hard
- 1 lane of HiSeq gives you a (bad?) genome
- you can quantify accurately against
very closely related spp. - Setaria viridis -> Setaria italica (>97%)
- Oryza rufipogon -> Oryza sativa (>95%)
WHAT'S A GOOD ASSEMBLY?
- contigs are realistic
- number (10k < n < 100k)
- length (50 < n < 30k)
- ORF% (60 < n < 95)
- supported by experimental evidence
- reads mapping properly
- not fragmented
- not chimeric
- makes sense in evolutionary context
- known proteins reconstructed
YOU CAN GET GOOD RESULTS

GARBAGE IN GARBAGE OUT
- sequence the right thing
- high-quality material
- check your reads

DON'T OVER-SEQUENCE
once you've sequenced ALL THE KMERS

everything new is error!
TAKE OUT THE GARBAGE: DISCARD AND TRIM
Trimmomatic


CONSERVATIVE IS NOT ALWAYS BETTER
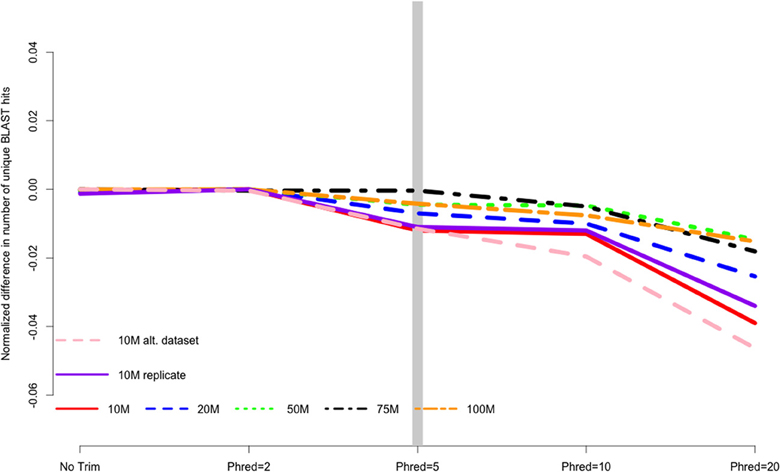
MacManes (2014) On the optimal trimming of high-throughput mRNA sequence data. Frontiers in Genetics
ERRORS AND BIOLOGICAL VARIATION: BAD
1 SNP

ERRORS AND BIOLOGICAL VARIATION: BAD
5 SNPs

VARIATION IS MEASURED BY HAMMING

Richard Hamming (1950)
VARIATION CAN BE ASSIMILATED

BayesHAMMER
assemblers have
complementary skill sets

PARAMETERS TOO

MANY ASSEMBLERS,
MANY PARAMETERS
- Velvet-Oases (k-sweep built in)
- TransAbyss (k-sweep built in)
- IDBA (iterative k-merging)
works great!
as long as you have a year to spare
high coverage is not useful


khmer diginorm (https://github.com/ged-lab/khmer)
READ SUBSETS ARE REPRESENTATIVE

THIS ALLOWS YOU TO OPTIMISE
- single assembly in ~30s
- 13,000 assemblies in a day
- choose the best areas of parameter space
fully automated optimisation:
ASSEMBLIES CAN BE MERGED
cluster (CD-HIT-EST)

sequence ID < 1 will collapse homeologs and variants
USE YOUR BIOLOGY
- don't trust small contigs
- real fusion transcripts are rare



RECOVERING VARIATION



OPTIMAL workflow
REMEMBER
- don't do transcriptomes if you don't have to
- design the experiment first
- many tissues, not too many reads
- inspect your reads at every stage
- clean, normalise
- check many parameter sets for multiple assemblers
- measure your assembly in a biologically meaningful way
- choose and merge the best
- you might be able to recover lost variation