How to study the entire brain
Daniel Fürth
Meletis Lab
DMC lab meeting
17th April 2015
daniel.furth@ki.se

- Rania
- Dinos
- Johanna
- Rickard
- Sissy
- Iakovos
Acknowledgement
Why studying the whole brain
- Population encoding of information.
- Concurrency.
- Functional anatomy is not the same as structural.
Why studying the whole brain
- Population encoding of information.


Corticospinal axons encode the direction and amplitude of muscle force rather than the direction of displacement. (Evarts, 1968)
Why studying the whole brain
- Population encoding of information. (Goergopoulos et al. 1982)

Why studying the whole brain
- Functional anatomy is not the same as structural. (Sanes et al. 1991)

Why studying the whole brain
- Functional anatomy is not the same as structural.
- WHAT IF: our current concept of modularity is totally wrong?
Do we really have a 'BigData' problem in neuroscience?
- Intel: Big data opportunities emerge in organizations generating a median of 300 terabytes of data a week. The most common forms of data analyzed in this way are business transactions stored in relational databases, followed by documents, e-mail, sensor data, blogs, and social media.
- 7 Tb full COLM brain = 43 brains a week!

I will define as big data anything that exceeds the size of a standard laptop hard drive. (Engert, 2014)
a mouse brain contains 500 x 10^9 cubic micron pixels, and if we want to record all of them for 20 min (1,000 s) at 1000 Hz, we again have 500 petabytes of raw data.
Do we really have a 'BigData' problem in neuroscience?
Do we really have a BigData problem in neuroscience?

Do we really have a 'BigData' problem in neuroscience?
Do we really have a BigData problem in neuroscience?

# find the location relative to the thunder installation
import os.path as pth
imagepath = pth.join(pth.dirname(pth.realpath(thunder.__file__)), 'utils/data/fish/tif-stack')
# load the images
data = tsc.loadImages(imagepath, inputformat='tif-stack')
import matplotlib.pyplot as plt
import seaborn as sns
sns.set_style('white')
sns.set_context('notebook')
img = data.first()[1]
plt.imshow(img[:,:,0], cmap="gray");
Freeman et al. (2014) Nat. Methods.
Do we really have a 'BigData' problem in neuroscience?
Do we really have a BigData problem in neuroscience?
# find the location relative to the thunder installation
import os.path as pth
imagepath = pth.join(pth.dirname(pth.realpath(thunder.__file__)), 'utils/data/fish/tif-stack')
# load the images
data = tsc.loadImages(imagepath, inputformat='tif-stack')
import matplotlib.pyplot as plt
import seaborn as sns
sns.set_style('white')
sns.set_context('notebook')
img = data.first()[1]
plt.imshow(img[:,:,0], cmap="gray");Freeman et al. (2014) Nat. Methods.
18534 x 27653 pixels
Do we really have a 'BigData' problem in neuroscience?
Twitter and Flickr have a 'BigData' problem: Tweets Flickr photos
The day before Christmas more photos are uploaded to Facebook than all photos on Flickr combined.
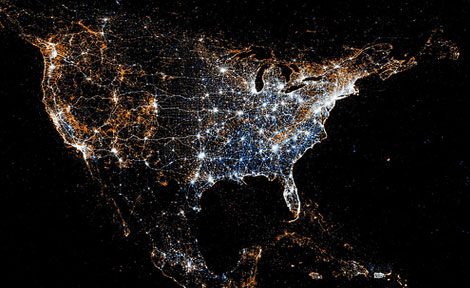


New York
Tokyo
Barcelona
New Orleans
Do we really have a 'BigData' problem in neuroscience?
Do we really have a 'BigData' problem in neuroscience?

http://www.parallac.org/
10 computers (146 processors)
Up to 64 cores per processor!
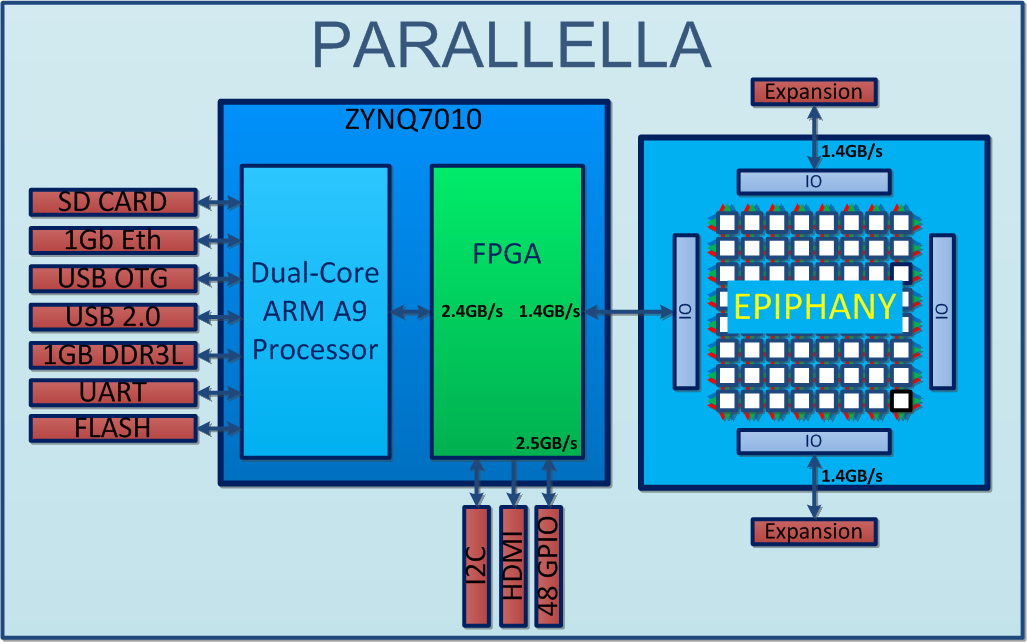

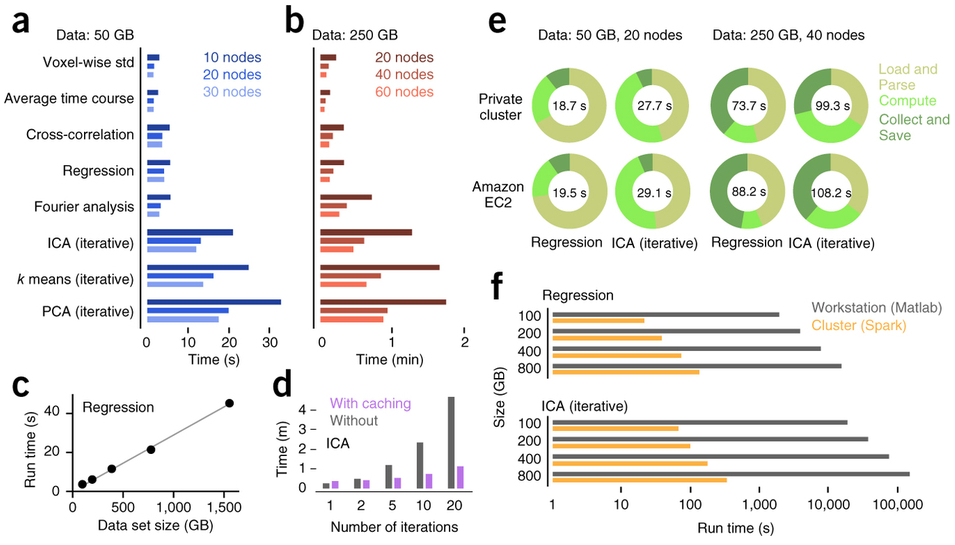
Freeman et al. (2014) Nature Methods
Results

Results

Whole-Brain Reconstruction
Pollak Dorocic et al. 2014


Reconstructing brain from sectioned tissue
Some problems with Allen's atlas
'Google maps' of neuroanatomy
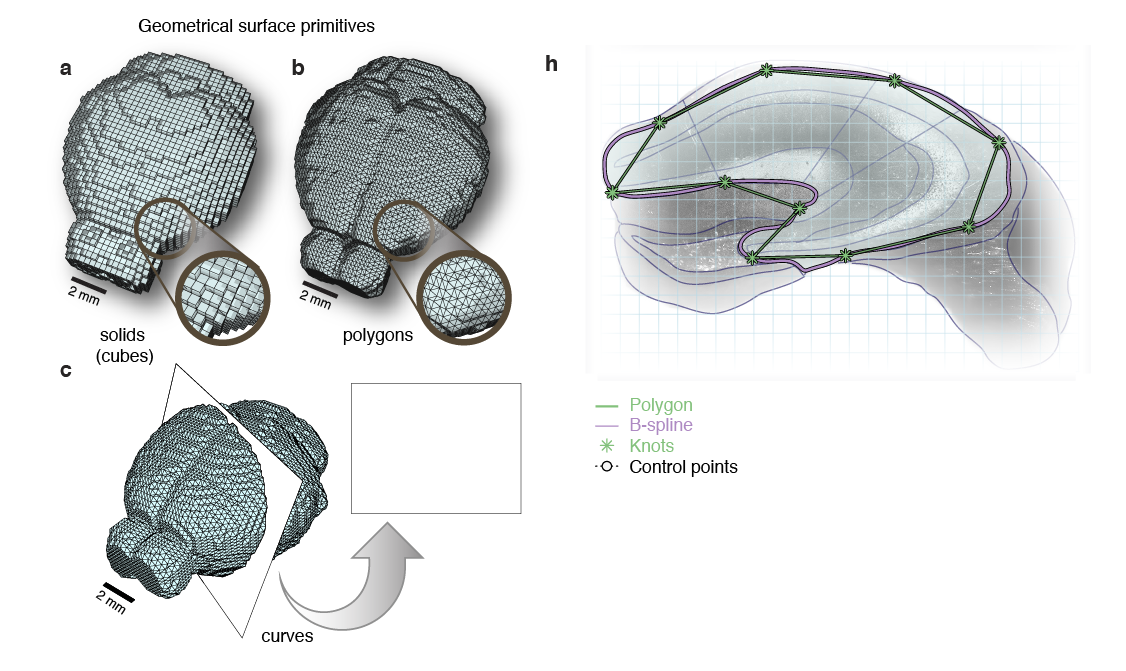
'Google maps' of neuroanatomy

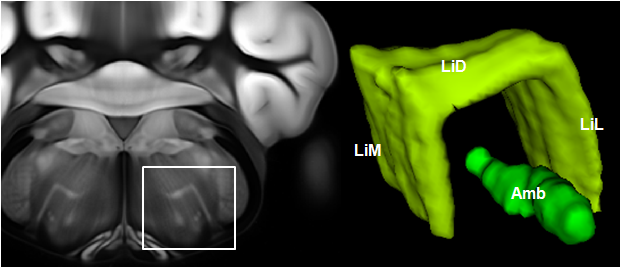
Non-Uniform Rational B-splines (NURBS)
'Google maps' of neuroanatomy
NURBS surface

'Google maps' of neuroanatomy

'Google maps' of neuroanatomy




similar to...
works with...
scRNA-seq
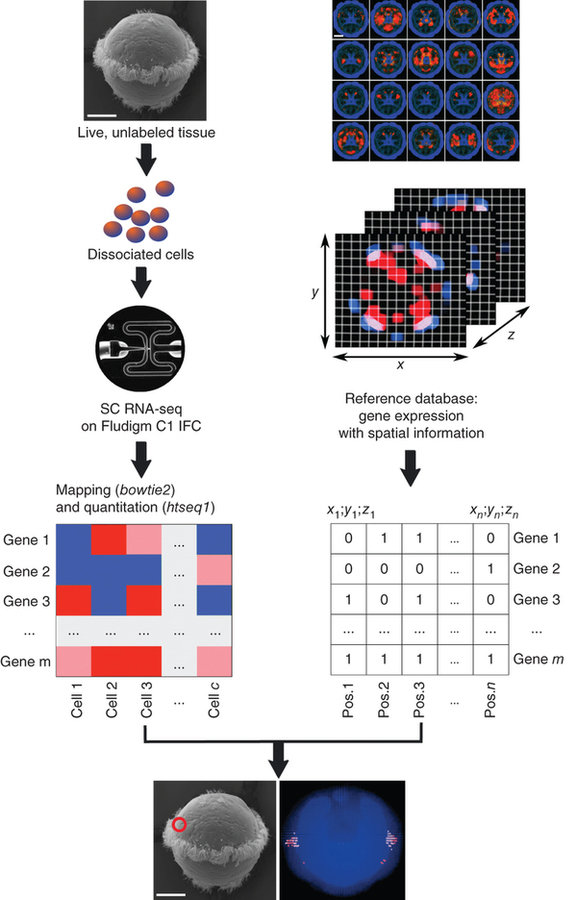
NATURE BIOTECHNOLOGY | COMPUTATIONAL BIOLOGY | ANALYSIS
High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin
Kaia Achim, Jean-Baptiste Pettit, Luis R Saraiva, Daria Gavriouchkina, Tomas Larsson, Detlev Arendt & John C Marioni
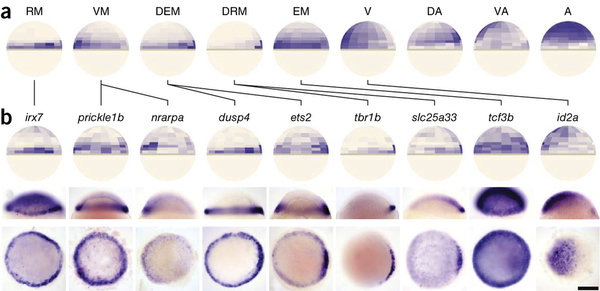
scRNA-seq
Allen Brain Reference Atlas

-
Atlas 2007 (manually drawn Nissl):
- 200 μm thick coronal sections.
-
Atlas 2011:
- 100 μm both coronal and sagital
- Atlas 2014 (connectivity avrg template)
-
Atlas 2015 (beginning of june):
- 10 x 50 μm
-
Registration atlas:
- 25 x 25 μm
-
Grid expression ISH:
- 200 x 200 μm MetaIOimage (.raw, .mhd)
scRNA-seq
Allen Brain Reference Atlas
scRNA-seq
scRNA-seq
Anatomic Gene Expression Atlas
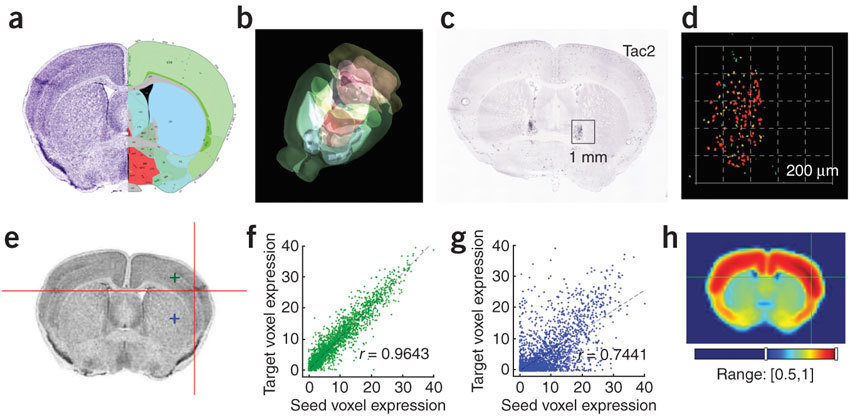
Lydia Ng, et al. (2009) Nat. Neuro.
http://mouse.brain-map.org/agea
scRNA-seq
LIM-homeodomain 6 (Lhx6)
General marker for cortical interneurons
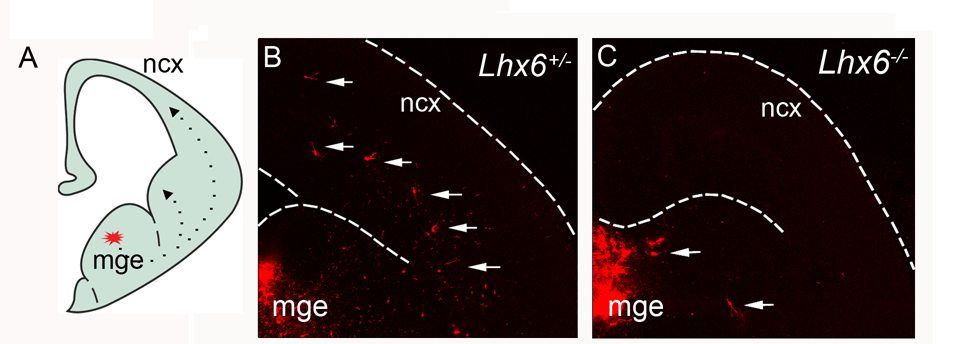
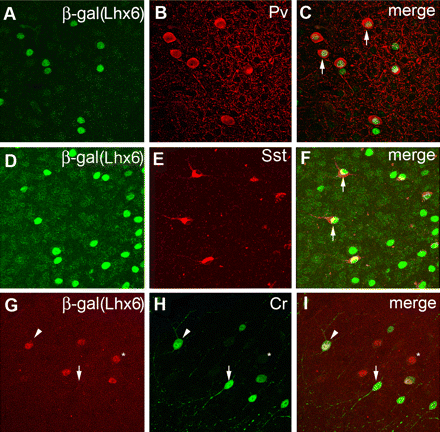
Migratory deficit of Lhx6-deficient MGE cells.
Liodis et al. 2007
scRNA-seq
LIM-homeodomain 6 (Lhx6)
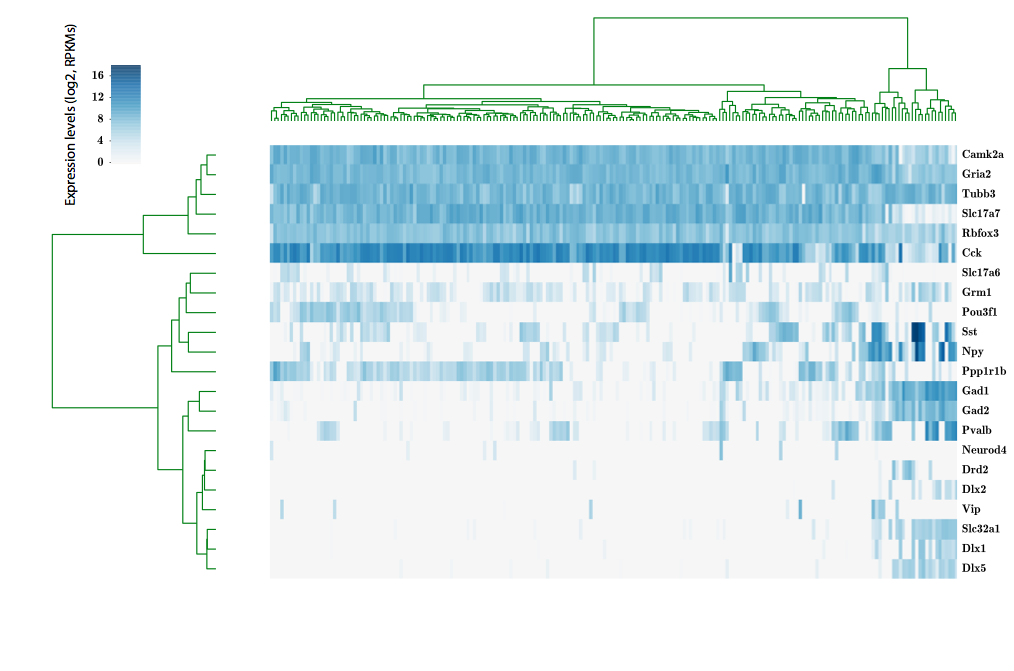
scRNA-seq
Our approach
scRNA-seq
Our approach
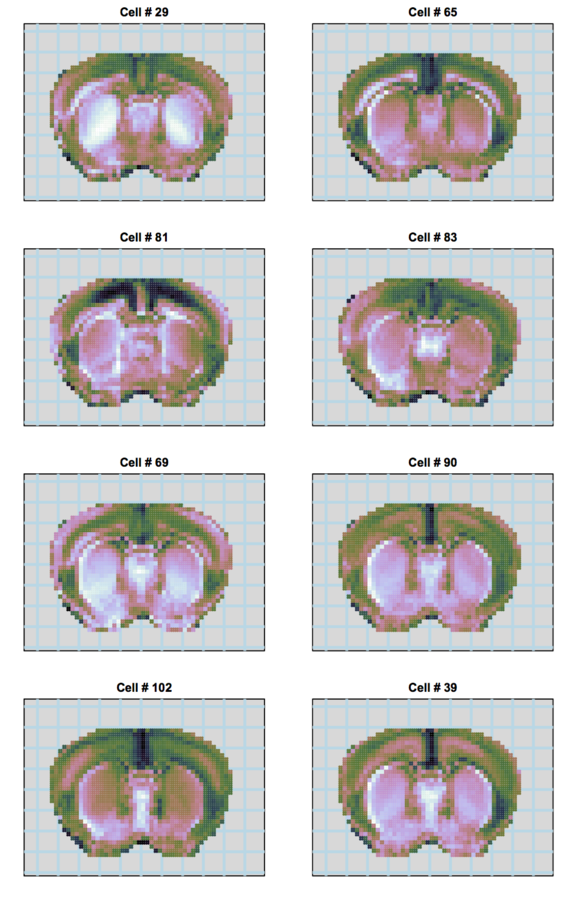
scRNA-seq
scRNA-seq
scRNA-seq
LIM-homeodomain 6 (Lhx6)
scRNA-seq
Allen Brain Reference Atlas

-
Atlas 2007 (manually drawn Nissl):
- 200 μm thick coronal sections.
-
Atlas 2011:
- 100 μm both coronal and sagital
- Atlas 2014 (connectivity avrg template)
-
Atlas 2015 (beginning of june):
- 10 x 50 μm
-
Registration atlas:
- 25 x 25 μm
-
Grid expression ISH:
- 200 x 200 μm MetaIOimage (.raw, .mhd)
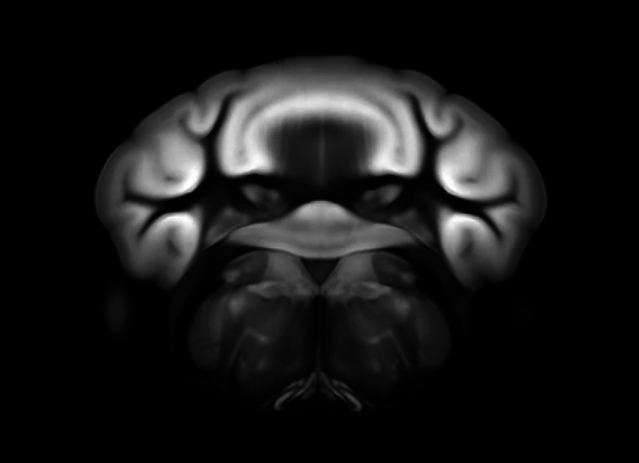
Connectivity average template (Ng et al. 2014)
scRNA-seq
Allen Brain Reference Atlas
-
Atlas 2007 (manually drawn Nissl):
- 200 μm thick coronal sections.
-
Atlas 2011:
- 100 μm both coronal and sagital
- Atlas 2014 (connectivity avrg template)
-
Atlas 2015 (beginning of june):
- 10 x 50 μm
-
Registration atlas:
- 25 x 25 μm
-
Grid expression ISH:
- 200 x 200 μm MetaIOimage (.raw, .mhd)
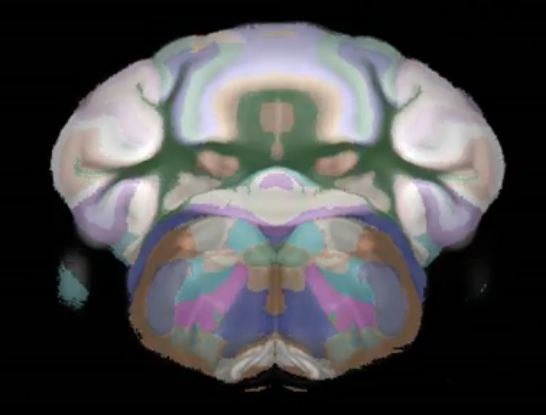
Connectivity average template (Ng et al. 2014)
Cocaine induced locomotoric activity
Cocaine induced locomotoric activity
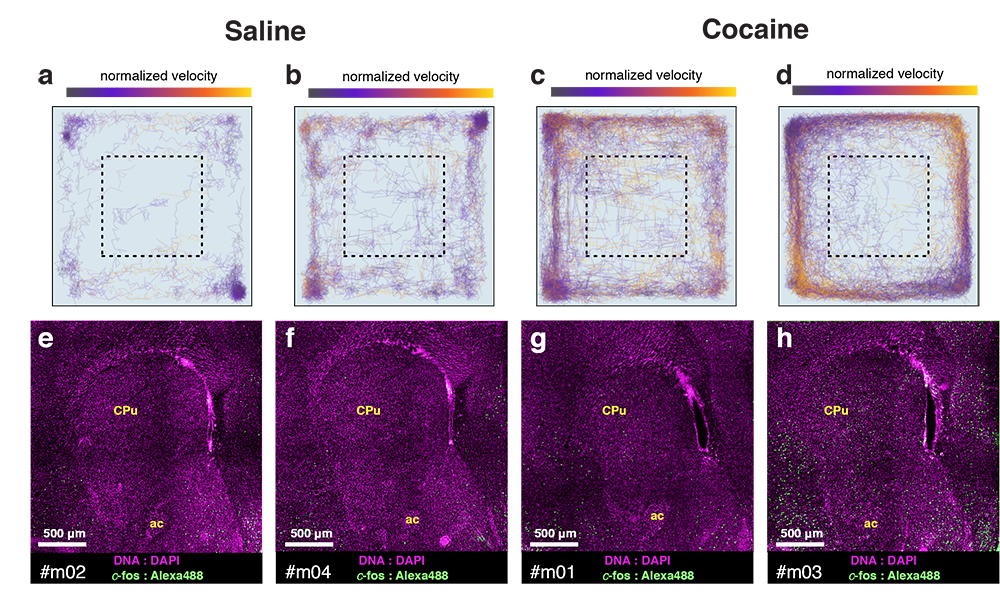
Whole-brain behavioral c-Fos mapping
Basic idea:
A
B
C
Independent
variable
Dependent
variable
Mediator
variable
Mediational statistical analysis
Whole-brain behavioral c-Fos mapping
Basic idea.
A
B
C
cocaine
dosage
(mg/ml)
Behavior
total track length (cm)
c-fos expression
(ith region)
direct effect
indirect effect
mediating effect
Question: How much of the behavioral variability is explained by variability in c-fos expression?
-
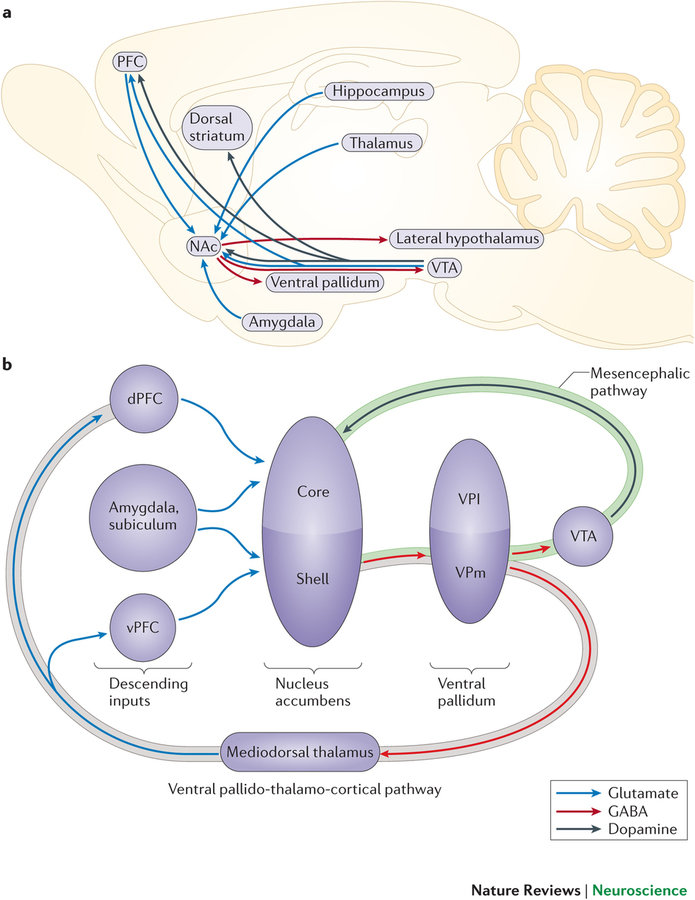
Regions of particular Interest
- NAc
- PFC
- Dorsal striatum
- Ventral pallidum
- Amygdala
- VTA
- LH
Kourrich, Calu & Bonci (2015)
Whole-brain behavioral c-Fos mapping
Whole-brain behavioral c-Fos mapping
Some problems with unspecific binding.
Whole-brain behavioral c-Fos mapping


Flat-field
Dark image




Whole-brain behavioral c-Fos mapping
Some problems with unspecific binding
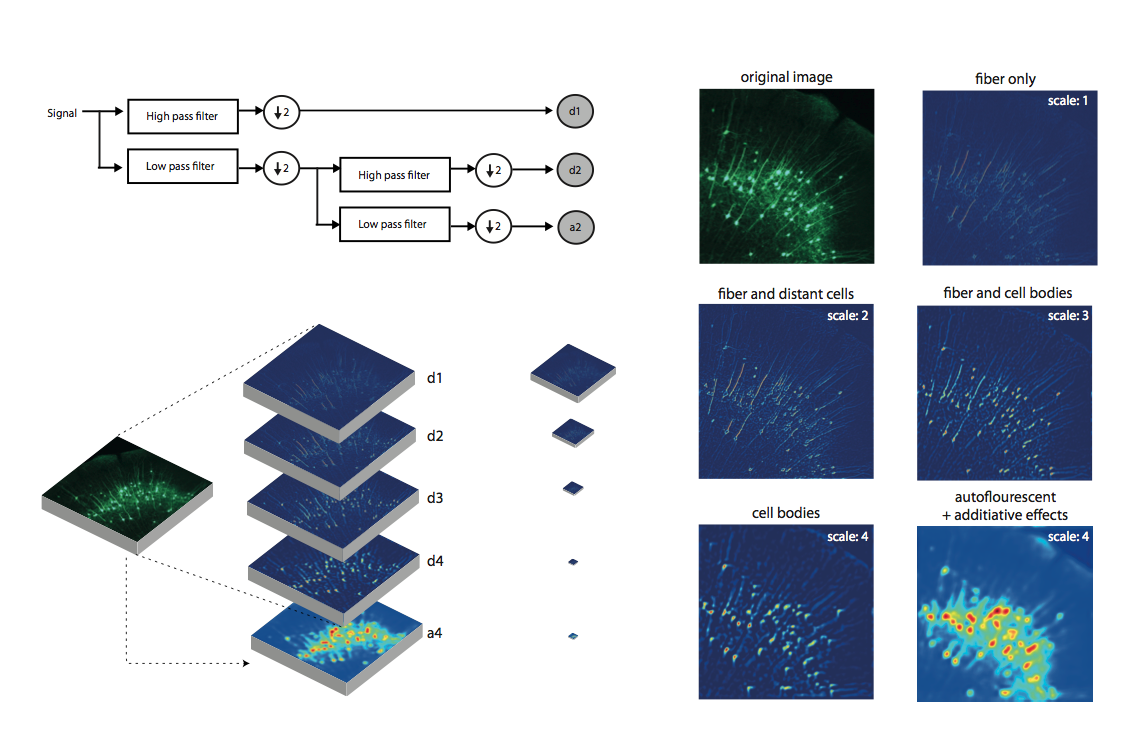
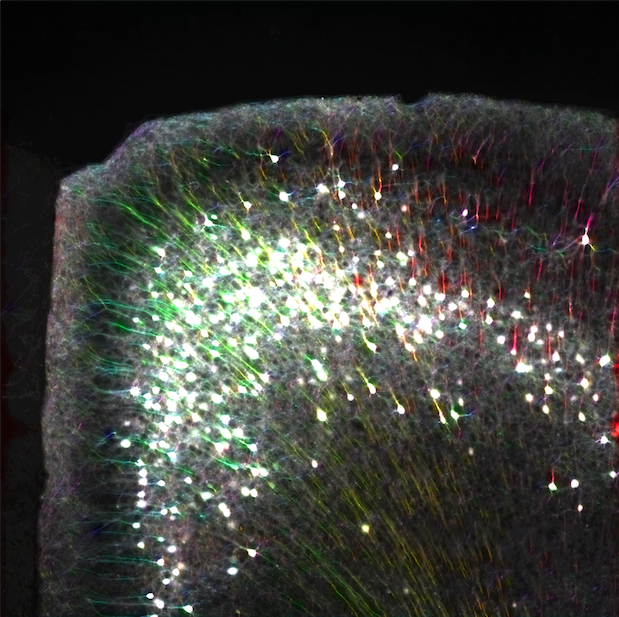
Whole-brain behavioral c-Fos mapping
Can be used to segment iut processes and their direction.
R package
- Why R?
- Standard data analysis:
- load some data
- estimate the density distribution.
- plot it
- Standard data analysis:
xx <- faithful$eruptions
fit <- density(xx)
plot(fit)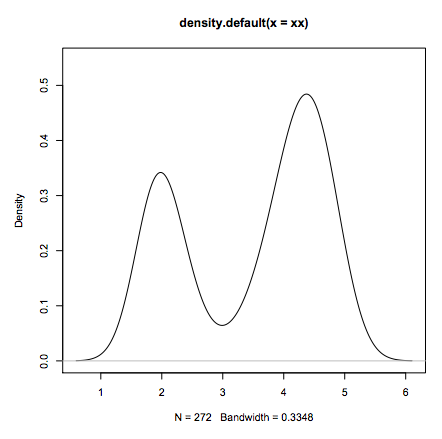
R package
- Why R?
#Line 1: loading
xx <- faithful$eruptions
#Line 2: estimate density
fit1 <- density(xx)
#Line 2: draw 10'000 bootstraps
fit2 <- replicate(10000, {
x <- sample(xx,replace=TRUE);
density(x, from=min(fit1$x), to=max(fit1$x))$y
})
#Line 3: compute 95% error "bars"
fit3 <- apply(fit2, 1, quantile,c(0.025,0.975))
#Line 4: plot the estimate
plot(fit1, ylim=range(fit3))
#Line 5: add estimation error as shaded region
polygon(c(fit1$x,rev(fit1$x)), c(fit3[1,], rev(fit3[2,])), col=’grey’, border=F)
#Line 6: add the line again since the polygon overshadows it.
lines(fit1)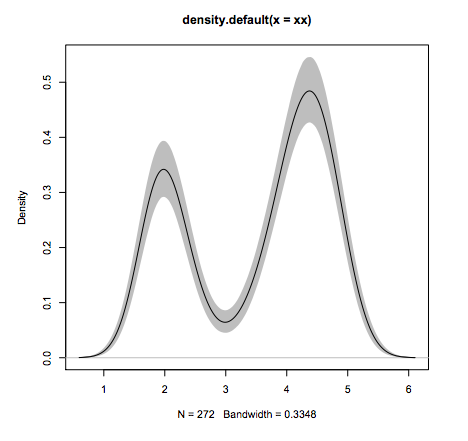
What other language can do this in 6 lines of code?
Parallel computing
- Parallel computing is extremely simple to implement from R.
# install.packages('foreach'); install.packages('doSNOW')
library(foreach)
library(doSNOW)
cl <- makeCluster(2, type = "SOCK")
registerDoSNOW(cl)
getDoParName()
#matrix operators
x <- foreach(i=1:8, .combine='rbind', .packages='wholebrain' ) %:%
foreach(j=1:2, .combine='c', .packages='wholebrain' ) %dopar% {
l <- runif(1, i, 100)
i + j + l
}Concurrency and parallel programming
- Multi threaded applications through .

#include <string>
#include <iostream>
#include <thread>
using namespace std;
//The functions we want to make the thread run.
void task1(string msg)
{
cout << "task1 says: " << msg;
}
void task2(string msg)
{
cout << "task1 says: " << msg;
}
//Main loop.
int main()
{
thread t1(task1, "Task 1 executed");
thread t2(task2, "Task 1 executed");
t1.join();
t2.join();
}Rcpp
Concurrency and parallel programming
- Multi-threaded applications through .
#include <string>
#include <iostream>
#include <thread>
using namespace std;
//The functions we want to make the thread run.
void task1(string msg)
{
cout << "task1 says: " << msg;
}
void task2(string msg)
{
cout << "task1 says: " << msg;
}
//Main loop.
int main()
{
thread t1(task1, "Task 1 executed");
thread t2(task2, "Task 1 executed");
//let main wait for t1 and t2 to finish.
t1.join();
t2.join();
}Rcpp
Dual core
Thank you!
scRNA-seq
Gene specificity
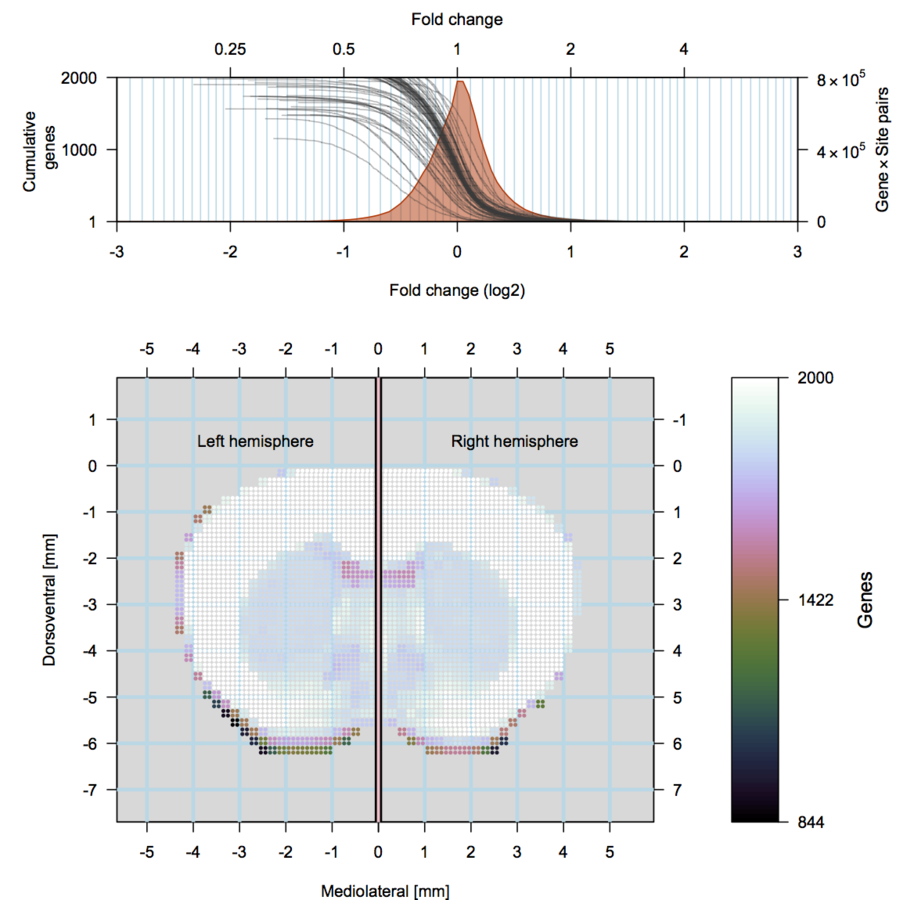
about ~24'000 genes expressed in the brain.
\text{Let us define the following variables.}
Let us define the following variables.
c : \text{a unique single cell.} \quad \text{Where: } c \in \{1, ... , C\}, \text{ and } C = 380.
c:a unique single cell.Where: c∈{1,...,C}, and C=380.
m : \text{a unique single gene.} \quad \text{Where: } m \in \{1, ... , M\}, \text{ and } C = 380.
m:a unique single gene.Where: m∈{1,...,M}, and C=380.
D : \text{a } C \times D \text{ read count matrix.}
D:a C×D read count matrix.
D_{c,m} : \text{ normalized number of reads mapped to cell } c \text{ for gene } m.
Dc,m: normalized number of reads mapped to cell c for gene m.
r_{c,m} : \text{ cell-gene specificity ratio.}
rc,m: cell-gene specificity ratio.
r_{c,m} = \frac{D_{c,m}}{ s_m }
rc,m=smDc,m
s_{m} = \frac{1}{C} \sum_{i=1}^C D_{i,m}
sm=C1∑i=1CDi,m
s_{m} : \text{ gene specificity.}
sm: gene specificity.
Whole-brain studies
By Daniel Fürth


