BLAST
Basic Local Alignment Search Tool

Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
- Los secuenciadores de última generación hacen que secuenciar organismos sea cada vez más "barato"
- El ensamblado y anotación de las secuencias continúa siendo una ardua tarea
- Ya hay mucha información de secuencias previamente anotadas
- No necesitamos partir de cero, se puede tratar de inferir información de anotaciones previas
- Para ello, existen muchas herramientas de búsqueda de secuencias similares a la nuestra
Búsqueda de similaridad en secuencias
- Las secuencias a analizar pueden ser fundamentalmente:
- Secuencias de proteínas (aminoácidos) (izqda.)
- Secuencias de ADN (nucleótidos) (dcha.)
Tipos de secuencias y búsquedas
Si dos secuencias son similares, muy probablemente...
- Deriven de una secuencia ancestral común
- Compartan una misma estructura
- Tengan una función biológica similar
Importancia de la similaridad
Homólogos (I)
- Dos secuencias muy similares
- ¿Qué significa muy similares? Bueno, puede haber discrepancias si tratamos de cuantificarlo, pero en varias fuentes aparecen:
- En proteínas, >25% aminoácidos idénticos
- En genes, >70% nucleótidos idénticos
- No tomándose como dogma, puede dar una idea
Homólogos (II)
- Para aportar mayor grado de certeza:
- E-valor (medida de significación estadística)
- Longitud de las secuencias
- Patrones de conservación
- Número de inserciones/borrados
Alineamiento de secuencias
- La búsqueda de similaridad se lleva a cabo mediante el alineamiento de secuencias
- Alineamiento de dos cadenas (entre dos dadas, o de una dada contra una base de datos)
- Global (secuencia completa contra otra/s)
- Local (busca subsecuencias similares)
- Semiglobal (huecos en extremos no penalizados)
- Alineamiento múltiple
Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
BLAST (I)
- Algoritmo más extendido de alineamiento local
- Método heurístico, no garantiza solución óptima
- Emplea programación dinámica
- Busca secuencias similares en bases de datos
- Existen muchos programas de la familia blast
- Existen muchas variantes de BLAST
- Y muchas herramientas para ello
BLAST (II)
- Funcionamiento básico
- Búsqueda de hits, subsecuencias similares cuya puntuación supere un umbral:
- En general, regiones idénticas de una cierta longitud. Al menos de alta puntuación en matriz de referencia (ej: BLOSUM)
- Extensión de pares de hits próximos (si tras la extensión superan umbral => HSP, high scoring pair)
- Evaluación del alineamiento (e-value)
- Búsqueda de hits, subsecuencias similares cuya puntuación supere un umbral:
BLAST (III) - Detalle
- Dadas dos secuencias, un par de segmentos es un par de subsecuencias de la misma longitud entre las secuencias problema y de la base de datos superando un umbral de puntuación.
- El algoritmo busca coincidencias de longitud fija, que se extienden a continuación hasta que se alcanzan ciertos parámetros umbral.
- Los pares de puntuación alta (HSP, high scoring pairs) constituyen la base de los alineamientos que obtiene como salida BLAST.
BLAST (IV) - Ejemplo
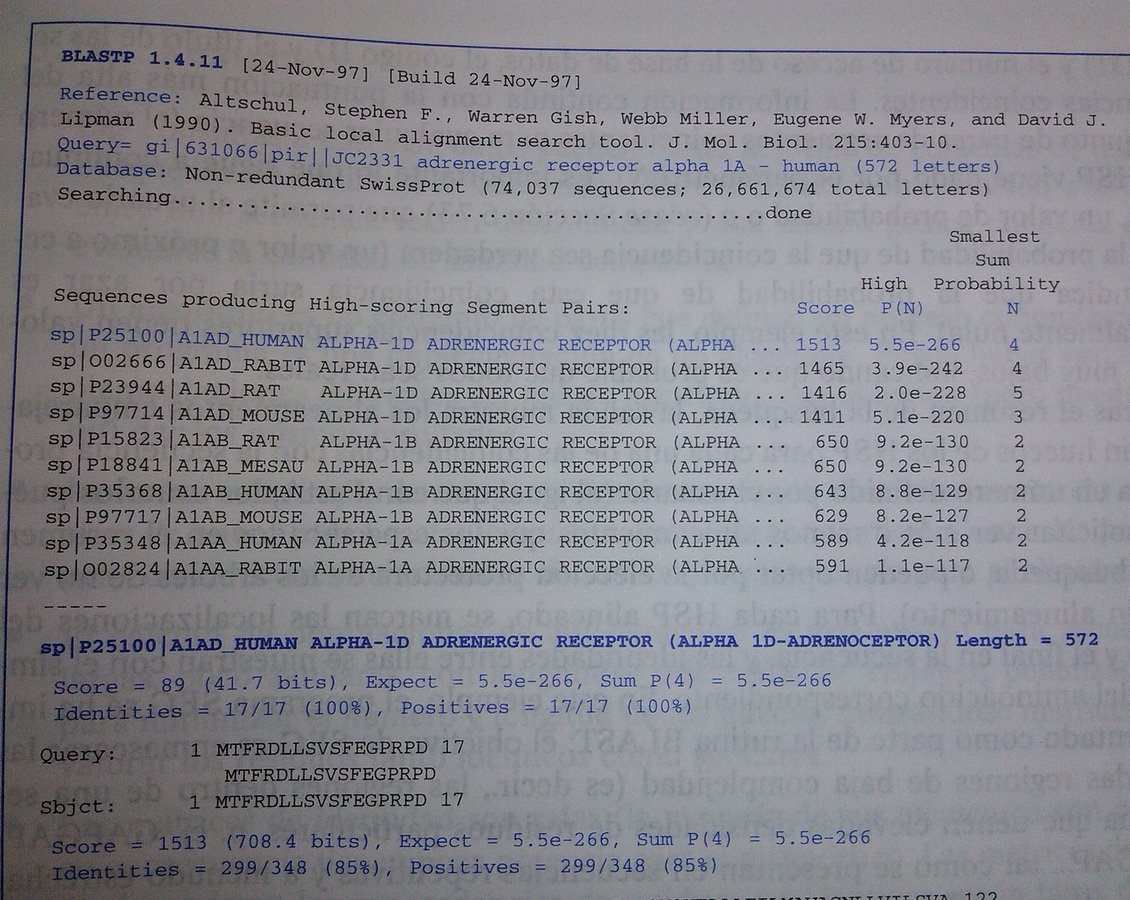
BLAST (V) - Descripción detallada
- Supongamos una secuencia Q y una base de datos D
- Dada una secuencia Q:
- Eliminar regiones de baja complejidad de Q
- Recopilar k-tuplas de Q
- Filtrar tuplas: mantener las de alta puntuación, por encima de un umbral T
- "Sembrar" un conjunto de posibles alineamientos
- Generar pares de alta puntuación (HSPs) a partir de las semillas
- Evaluar la significancia/importancia estadística de las coincidencias de los HSPs
- Presentar el informe de alineamientos encontrados a partir de los HSPs
BLAST. 1-Eliminar baja complejidad
- Si intentáramos alinear HHHHHHHHKMAY con HHHHHHHHURHD:
- Tendrá alta puntuación en matriz BLOSUM
- Pero la parte significativa es KMAY vs URHD
- Por tanto, eliminaremos la región de H's (de baja complejidad) de la secuencia Q
-
El módulo SEG que suele venir con BLAST hace esa labor, aunque puede desactivarse.
- Idea: establece una ventana que se mueve en Q, usa una rutina para determinar baja complejidad, y reemplaza el segmento por algo simplificado
BLAST. 2-Recopilar k-tuplas en Q
-
k la longitud de la ventana con la que recorrer Q:
- Generalmente 3 para proteínas, 11 para ADN
- Puede variar
- Las tuplas suelen llamarse palabras (w, words). Al conjunto de las mismas se las suele denominar W
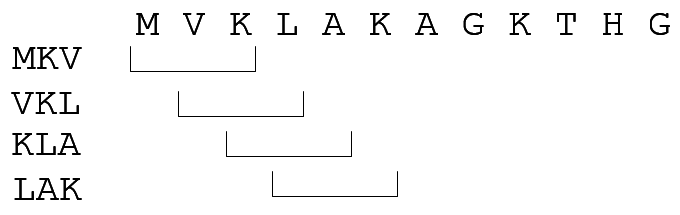
BLAST. 3-Encontrar k-tuplas de alta puntuación
- Dada una palabra w de W, encontrar en la base de datos otras palabras de longitud k tales que:
- Aparezcan en alguna secuencia, o bien
- Su puntuación según la matriz (BLOSUM o similar) supere un umbral T
- Elegir T para limitar el número considerado
- Llamar a esas palabras tuplas de alta puntuación
BLAST. 3-Encontrar k-tuplas de alta puntuación - Ejemplo
- Ej: sea w=MVK, y sea T=8
-
Supongamos que encontramos MVK, MEK, PVK, MVL y FVK en la base de datos
-
Blosum(MVK,MVK) = 5+4+5=14
-
Blosum(MVK,MEK) = 5-2+5=8
-
Blosum(MVK,PVK) = -2+4+5=7
-
Blosum(MVK,MVL) = 5+4-2=7
-
Blosum(MVK,FVK) = 0+4+5=9
-
-
Sólo se mantendrán MVK y FVK
BLAST. 3-Encontrar k-tuplas de alta puntuación - Consideraciones
-
Para cada w en W, se habrán encontrado todas las tuplas de alta puntuación.
- Se deben organizar, recordando todos los lugares de Q donde se encontraba.
-
Cada tupla de alta puntuación será una semilla
-
Para generar posibles alineamientos
-
Una semilla puede generar más de un alineamiento
-
BLAST. 4-"Sembrar" posibles alineamiento
-
Para cada tupla v en Q (no en W):
- Recuperar las palabras de alta puntuación, Hv.
- Recuperar la lista de lugares de Q donde ocurre v, Pv.
-
Para cada par (w en Hv, pos en Pv)
-
Encontrar todas las secuencias (D) de la base de datos:
-
Con una coincidencia exacta con w en pos'.
-
-
Almacenar un alineamiento entre Q y D, con v en la posición pos en Q y w en la posición pos' en D
-
BLAST. 4-"Sembrar" posibles alineamiento - Ejemplo
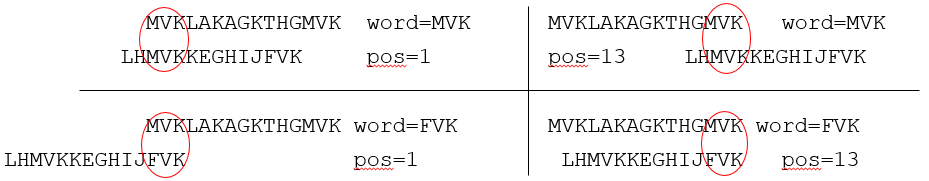
- Supongamos Q = MVKLAKAGKTHGMVK
- Comenzaríamos con v=MVK:
- Hv = {MVK, FVK}, Pv = {1, 13}
- Supongamos la secuencia en la base de datos
- D = LHMVKKEGHIJFVK
- Los posibles alineamientos "sembrados" serían:
BLAST. 5-Generar pares de alta puntuación (HSPs)
-
Para cada "alineamiento" A, con secuencias Q y D emparejadas, con la región original de emparejamiento M:
- Extender M a la izquierda, hasta que la puntuación comience a decrecer
- Extender M a la derecha, hasta que la puntuación comience a decrecer
- De los tramos de secuencias mayores que la tupla original, nos quedamos con los de puntuación mayor que un umbral de puntuación, y deshechamos el resto
BLAST. 5-Generar pares de alta puntuación (HSPs) - Ejemplo

Así, tras la extensión por la derecha, nos quedamos con MVKLQT, con puntuación 21
BLAST. 6-Comprobar significancia estadística
- Se trató de extender la regiones a alinear, para minimizar la probabilidad de que la coincidencia o la puntuación según la matriz (BLOSUM, etc.) ocurra por azar
- Cuestión a dilucidar: ¿es un HSP significativo?
- Supongamos un HSP con puntuación S para una región de longitud L en las secuencias Q y D
- Se calcula la probabilidad de que dos secuencias aleatorias Q' y D' puntúen S en una región de longitud L, con Q' de igual longitud que Q y D' que D.
- Esta probabilidad deberá ser muy pequeña para que sea significativo el HSP
BLAST 7 Informar de los alineamientos
- Por cada HSP estadísticamente significativo:
- Se incluye el alineamiento en el informe
- Si una secuencia D tiene dos HSPs con la secuencia Q:
- Se incluyen en el informe dos alineamientos distintos
- Algunas versiones de BLAST unifican ambos.
Variantes de la comparación
- blastp: proteínas con base de datos de proteínas
- tblastn: proteínas con bd nucleótidos
- blastn: nucleótidos con base de datos de nucleótidos
- blastx: nucleótidos con base de datos de proteínas
- tblastx: a partir de nucleótidos traduce a proteínas, que compara con base de datos de nucleótidos
BLAST. Capacidades y alternativas
- Encontrar genes en un genoma: subsecuencias del genoma, blastx contra bd NR, o bien ejecutar software de predicción de genes más complicado
- Predecir la función de una proteína: lanzar blastp o blastx contra swissprot, o conducir experimentos de análisis de dominio en laboratorio
- Predecir estructura 3-D de proteínas: blastp contra bd PDB, o conducir modelización de homología, o análisis por rayos X o NMR de las proteínas
- Encontrar miembros de la familia: blastp o PSI-blast contra NR, y multiple sequence alignment, o bien clonar nuevos miembros usando técnicas de PCR
Herramientas para lanzar BLAST
- Aplicaciones web
- Aplicaciones de escritorio/línea de comandos
- Bibliotecas software
Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
Herramientas online
Podemos lanzar BLAST en:
- Web de NCBI - BLAST
(National Center for Biotechnology Information)
- Sequece similarity search (sss) en EBI
(European Bioinformatics Institute)
(Swiss Inst. of Bioinformatics, EMBnet server)
- Sanger BLAST, Ensembl, UCSC, DDBJ, etc.
BLAST para secuencias proteicas
- Objetivo: tenemos una secuencia proteica, y queremos encontrar otras similares en una base de datos de secuencias.
- Dos opciones:
- blastp: proteínas con base de datos de proteínas (para descubrir algo sobre mi proteína)
- tblastn: proteínas con base de datos de nucleótidos (para descubrir nuevos genes que codifiquen proteínas simples)
- En caso de duda, usar blastp
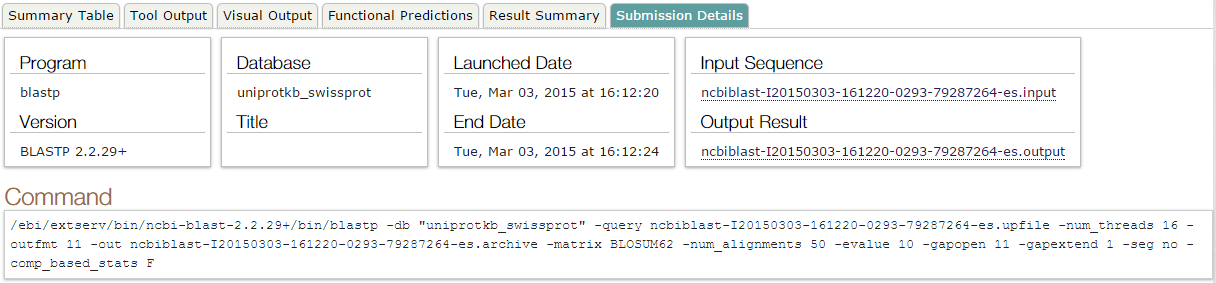
blastp en NCBI. Ejecución (I)
- Acudimos al servidor de NCBI para lanzar pblast
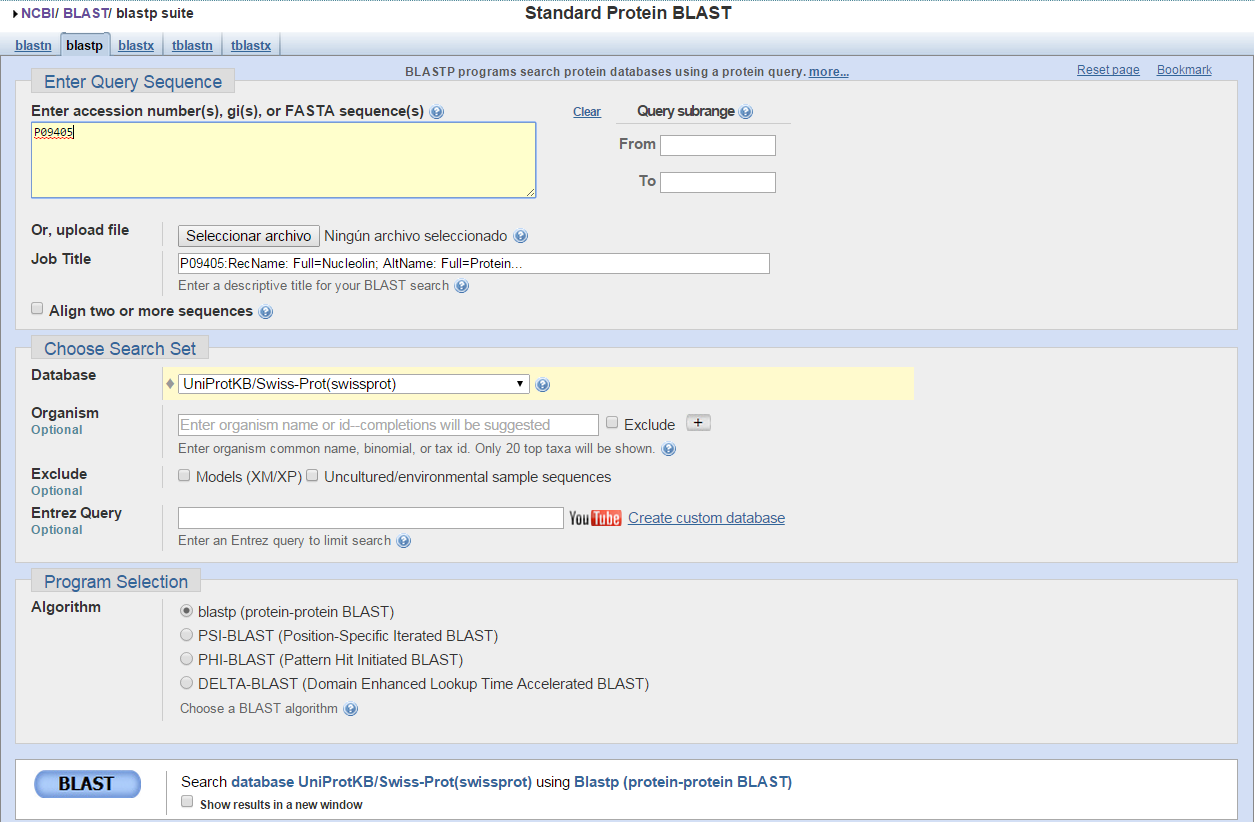
blastp en NCBI. Ejecución (II)



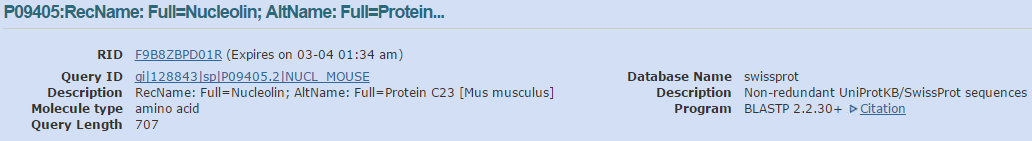
blastp en NCBI. Resultados (I)
blastp en NCBI. Resultados (II)
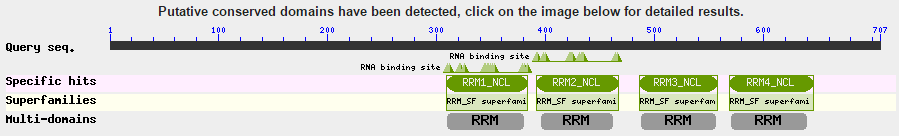
blastp en NCBI. Resultados (II')
- Puntuación
- Query cover (% de la secuencia buscada que se alinea con la secuencia encontrada)
- Ident (% de similaridad entre ambas secuencias sobre la longitud del área cubierta)
- E-value (nº hits que uno podría esperar ver por azar; decrece exponencialmente conforme se incrementa la puntuación del alineamiento)
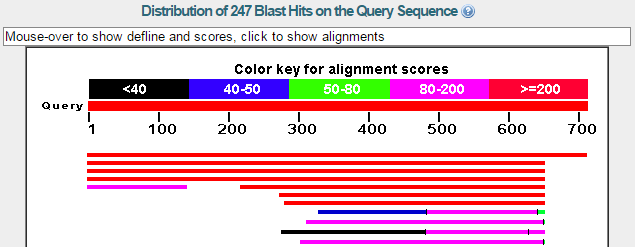
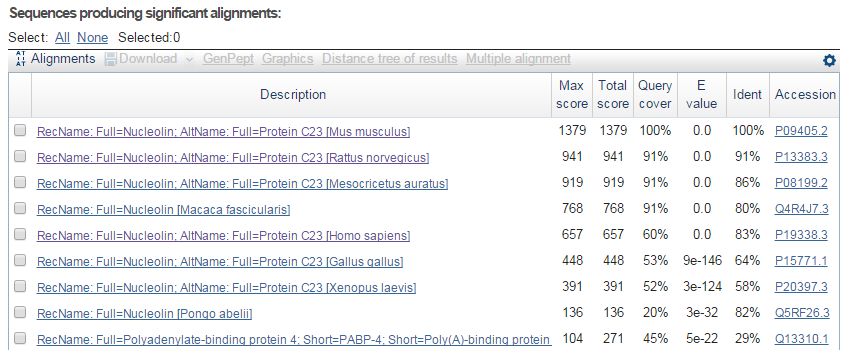
blastp en NCBI. Resultados (III)
- Alineamientos más significativos:
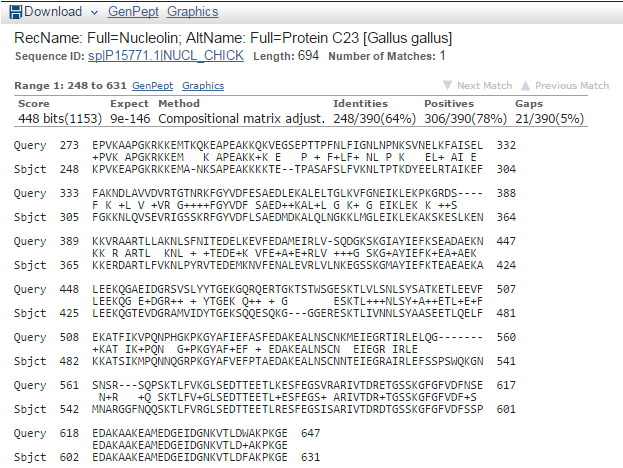
blastp en NCBI. Resultados (IV)
- Detalle del alineamiento (desde la vista gráfica o la lista de alineamientos + significativos):
blastp en NCBI. Conservar datos (I)
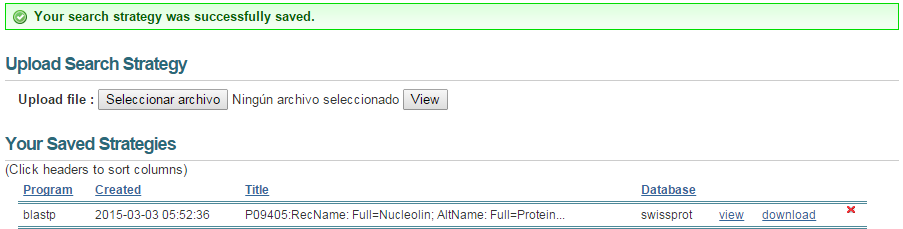

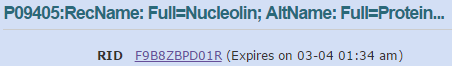
blastp en NCBI. Conservar datos (II)
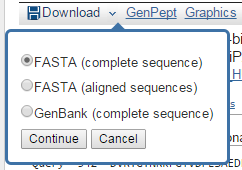
- Descargar resultados:
- Globales:
- Individuales:

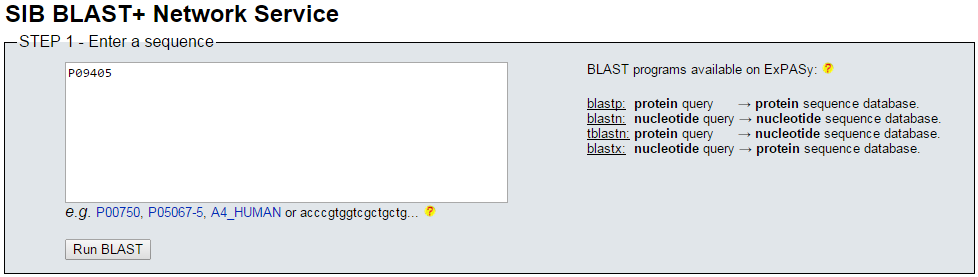
blastp en ExPASy (I)
- Acudimos a la web de ExPASy para lanzar blastp, e introducimos el mismo accession number, P09405, con la selección de base de datos de proteínas, y hacemos Run.
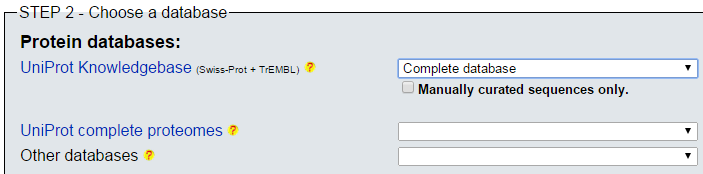
blastp en ExPASy (II)
- Visualizamos los resultados gráficos:
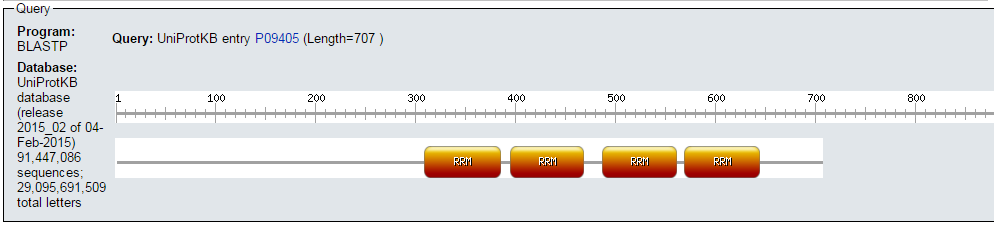
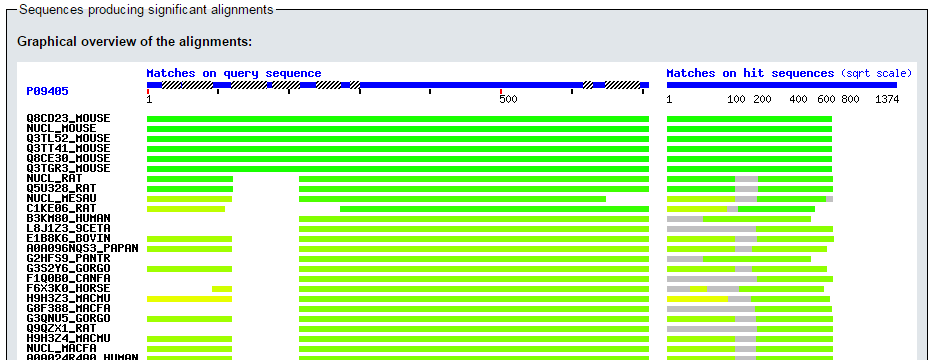
blastp en ExPASy (III)
- Así como la lista de hits:
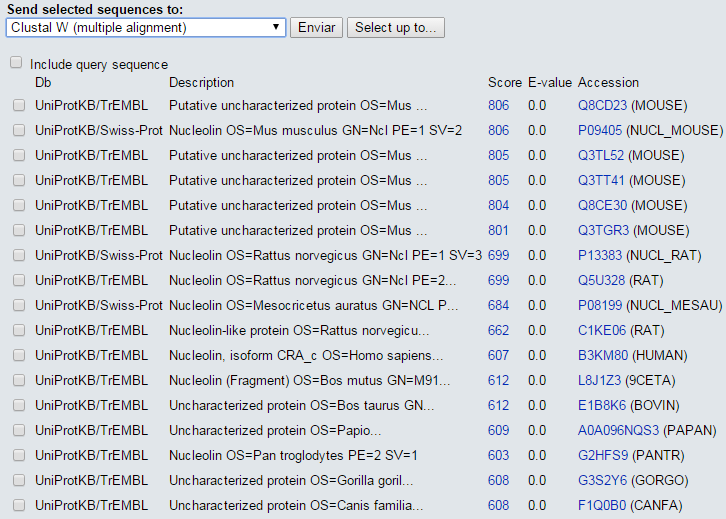
blastp en ExPASy (IV)
- Detalle de un alineamiento:
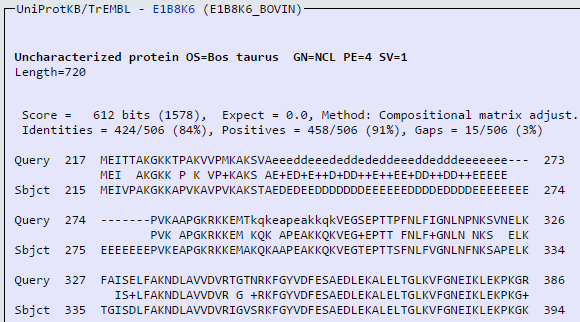
blastp en ExPASy (V)
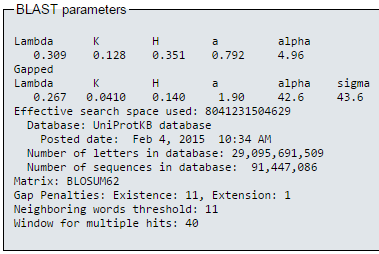
blastp en EBI (I)
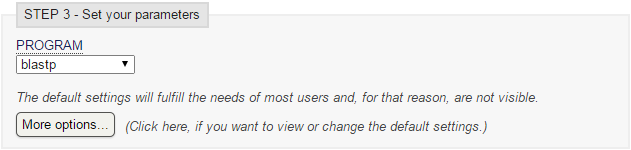
- Vamos al servidor del EBI, seleccionamos Swiss-Prot, seleccionamos archivo de secuencia, y programa blastp:
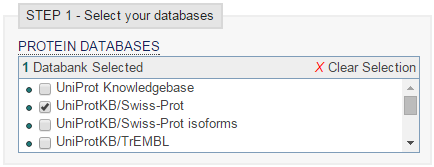
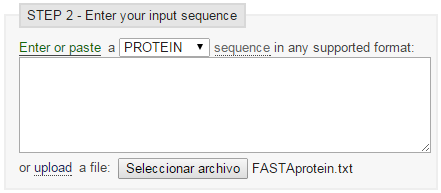
blastp en EBI (I')
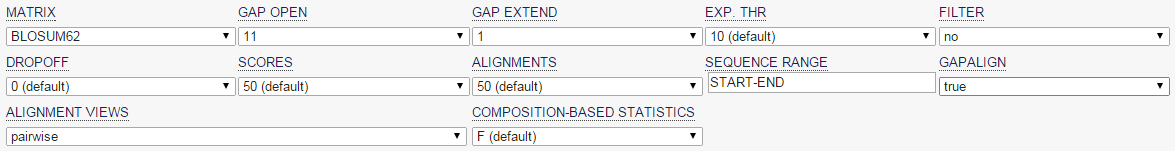
blastp en EBI (II)
- Obtenemos la lista de hits como salida:
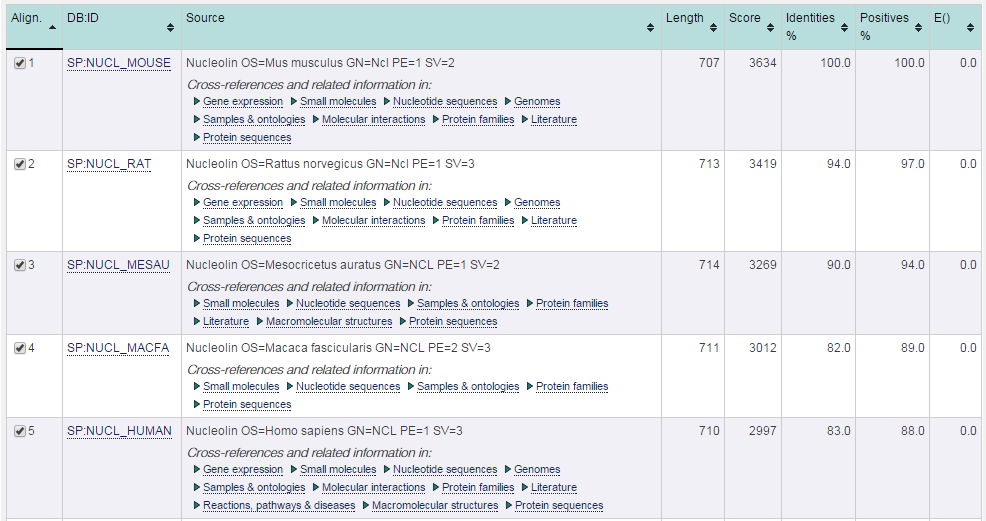
blastp en EBI (III)
- Podemos ver detalle de alineación o anotación:
- Como vemos, también podemos descargar datos
o enviar a otra herramienta
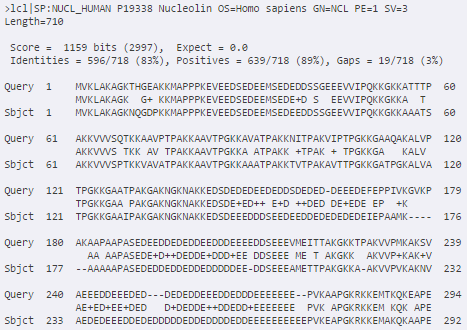

blastp en EBI. Salidas adicionales (I)
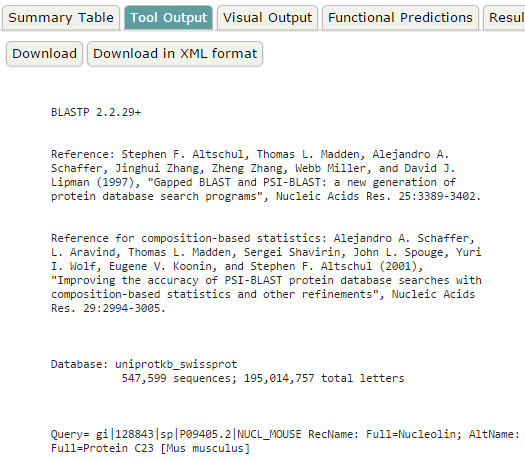
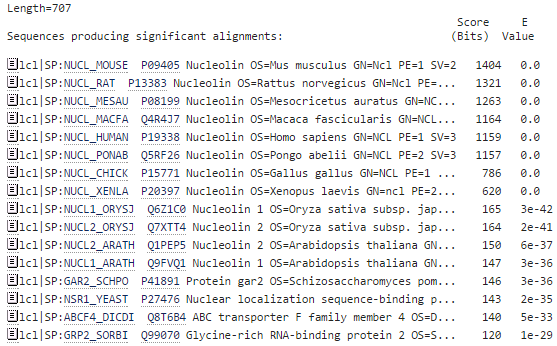
blastp en EBI. Salidas adicionales (II)
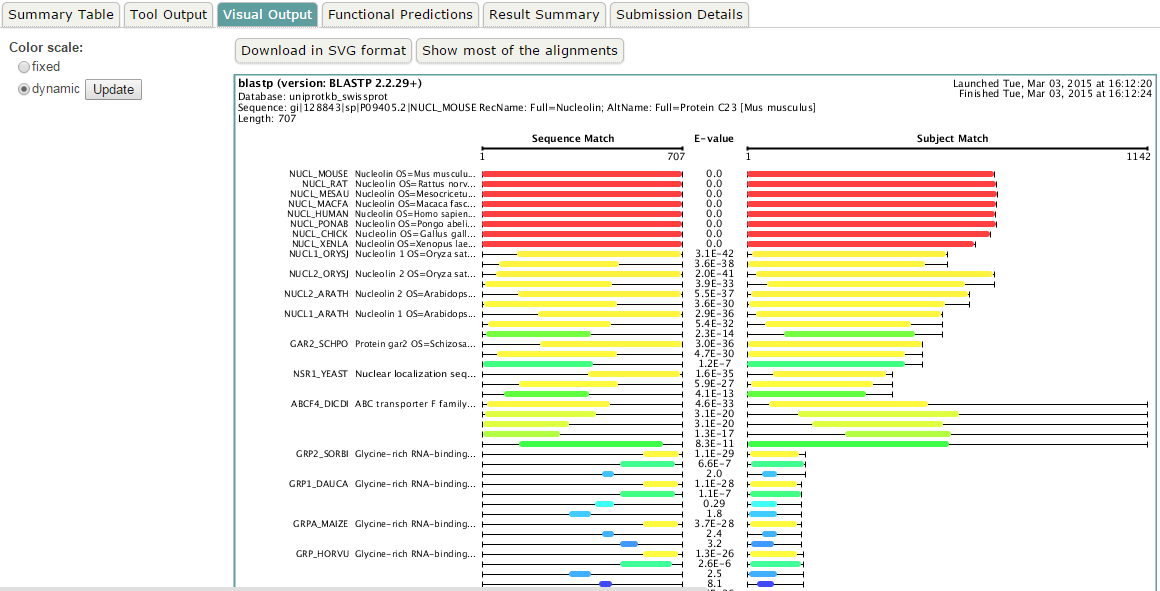
blastp en EBI. Salidas adicionales (III)
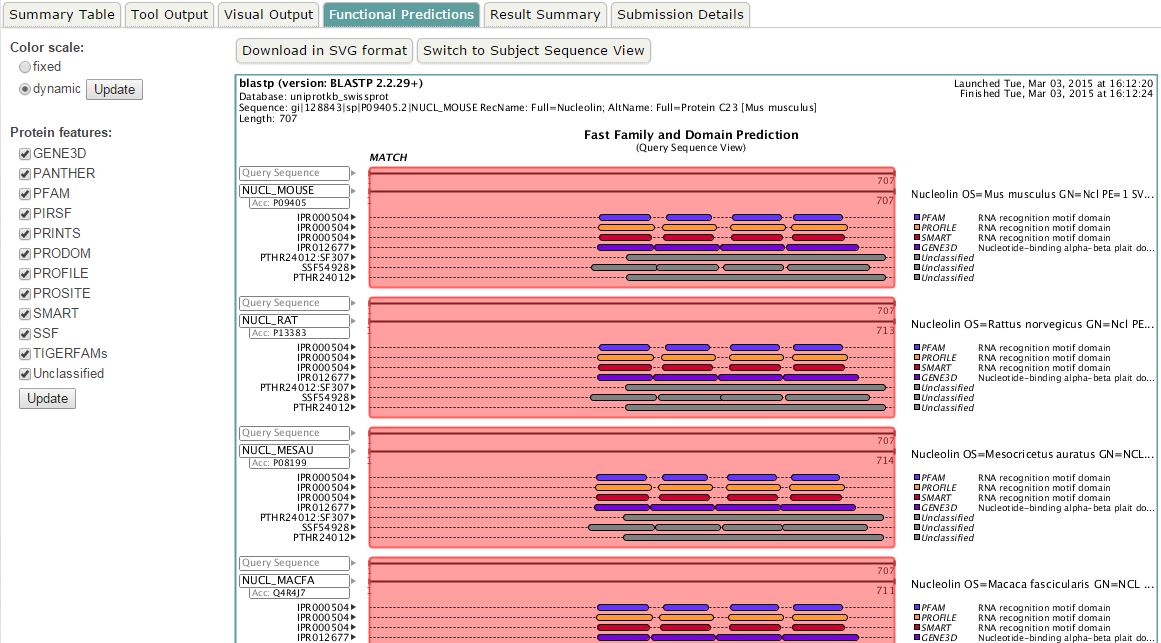
blastp en EBI. Salidas adicionales (IV)
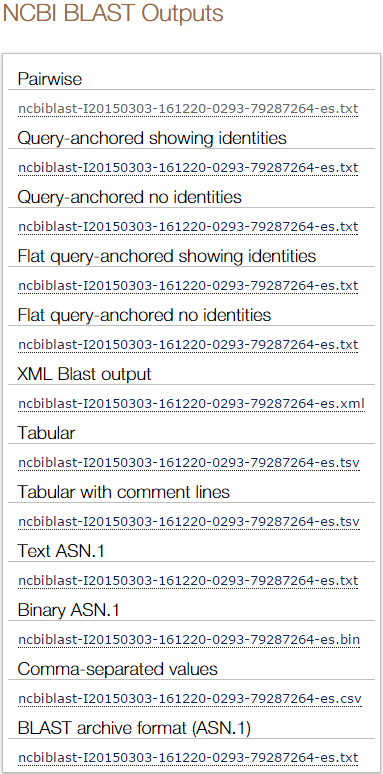
blastp en EBI. Salidas adicionales (V)


Otros servidores
BLAST para secuencias nucleotídicas
- Los principios son los mismos que en las anteriores
- Requiere operaciones similares
- No es tan eficiente como en el caso de las proteínas
BLAST para ADN. Objetivos
- Objetivo: tenemos una secuencia nucleotídica, y queremos encontrar otras similares en una base de datos de secuencias
- Tres opciones:
- blastn: secuencias de nucleótidos contra bd de nucleótidos, para secuencias muy similares
- tblastx: secuencias recibida y encontrada traducidas a proteínas
- blastx: secuencia recibida contra bd de secuencias proteicas
BLAST ADN. ¿Cuál usar?
- Si estoy interesado en ADN no codificante, blastn
- Si quiero descubrir nuevas proteínas, tblastx
- Si quiero descubrir proteínas que están codificadas en mi cadena de ADN, blastx
- Si no estoy seguro de la calidad de mi cadena de ADN (puede tener errores de secuenciación), blastx
BLAST ADN. ¿Qué bd y criterio?
- A menos que estemos usando blastx (que se lanza contra bd de proteínas), debemos selecionar una base de datos de secuencias nucleotídicas
- Restringe la búsqueda a bd más específica, ya que estas búsquedas son más lentas
- Establece criterios más restrictivos de filtrado si es necesario
blastn en NCBI. Ejercicio
- Con lo que ya hemos visto para las proteínas, podemos realizar este ejercicio:
- Acceder al servidor de NCBI
- Determinar si esta secuencia de ADN que hemos obtenido en un secuenciador tiene similaridad con algunos genes conocidos, para poder consultar anotaciones
- Quedarnos con la primera secuencia cuyo gen tenga RefSeq a revisado (accession NM_...)
- ¿Cómo se llama el gen? ¿Podemos en principio considerarlo homólogo?
blastn en NCBI. Ejercicio resuelto
- Los detalles de este ejercicio se pueden encontrar en:
-
Solución detallada
-
Solución detallada
Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
BLAST+ para Unix
Distintas formas de obtenerlo:
- Paquete ncbi-blast+ en Ubuntu
- Descarga e instalación manual
Una vez instalado, podemos descargarnos bases de datos como indica el manual de arriba.
En este enlace podemos ver el listado de bases de datos disponibles.
BLAST+ para Unix - Ejercicio
Podemos emplear estas utilidades con el mismo ejemplo de proteínas visto, mediante:
-
ftp ftp.ncbi.nlm.nih.gov, usuario anonymous
- cd blast/db
- bin
- get swissprot.tar.gz
- exit
- tar zxvpf swissprot.tar.gz
- makeblastdb -entry P09405 -db swissprot > P09405.fa
- blastp -query P09405.fa -db swissprot -out P09405.blast
Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
Perl
- Existen muchas bibliotecas y scripts para poder trabajar con BLAST desde distintos lenguajes de programación, para:
- Automatizar tareas
- Pre-procesar/post-procesar secuencias o alineamientos
- ...
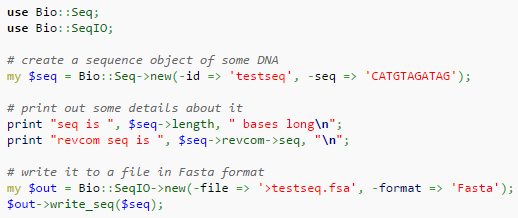
- Perl es un buen ejemplo de ello.
- Manual introducción a Perl
Perl
Python
- Otro lenguaje de programación con muchas bibliotecas y recursos para bioinformática es Python
- Tutorial introducción a Python
- Biopython incluye numerosos recursos para bioinformática.
- En particular, permite también trabajar con BLAST
Python


Python
- Un ejemplo simple para guardar solamente los primeros diez alineamientos:
- Más sobre el parsing de la salida aquí

Introducción
BLAST
Herramientas online
Herramientas de escritorio
Bibliotecas
Trabajos propuestos
Trabajos propuestos
- Estudiar las distintas variantes de BLAST que existen en la actualidad, incluyendo opciones con huecos, PSI-BLAST, etc.
- Estudiar alternativas a BLAST, tanto exhaustivas como heurísticas, detallando las similitudes y diferencias, ventajas e inconvenientes
- Explorar el uso de BLAST a través de bibliotecas de lenguajes de programación como:
- Python
- Perl
- R
Bibliografía
- La bibliografía empleada en esta presentación incluye los recursos detallados en este documento.
- También han sido de utilidad las fuentes referenciadas mediante enlaces en estas dispositivas, y algunos libros adicionales:
blast
By Luis Valencia Cabrera