Computational Biology Seminar
Class 05 - Storytelling
(BIOSC 1630)
September 27, 2023
Towards Atomistic Modeling of Complex Environments with Many-Body Machine Learning Potentials
Alex M. Maldonado
aalexmmaldonado
Maldonado, A. M.; et al. Digital Discovery 2023, 2, 871-880. DOI: 10.1039/D3DD00011G
Clean energy is growing, but slowly
Solvation plays an important role in advancing energy technologies
Nuclear power
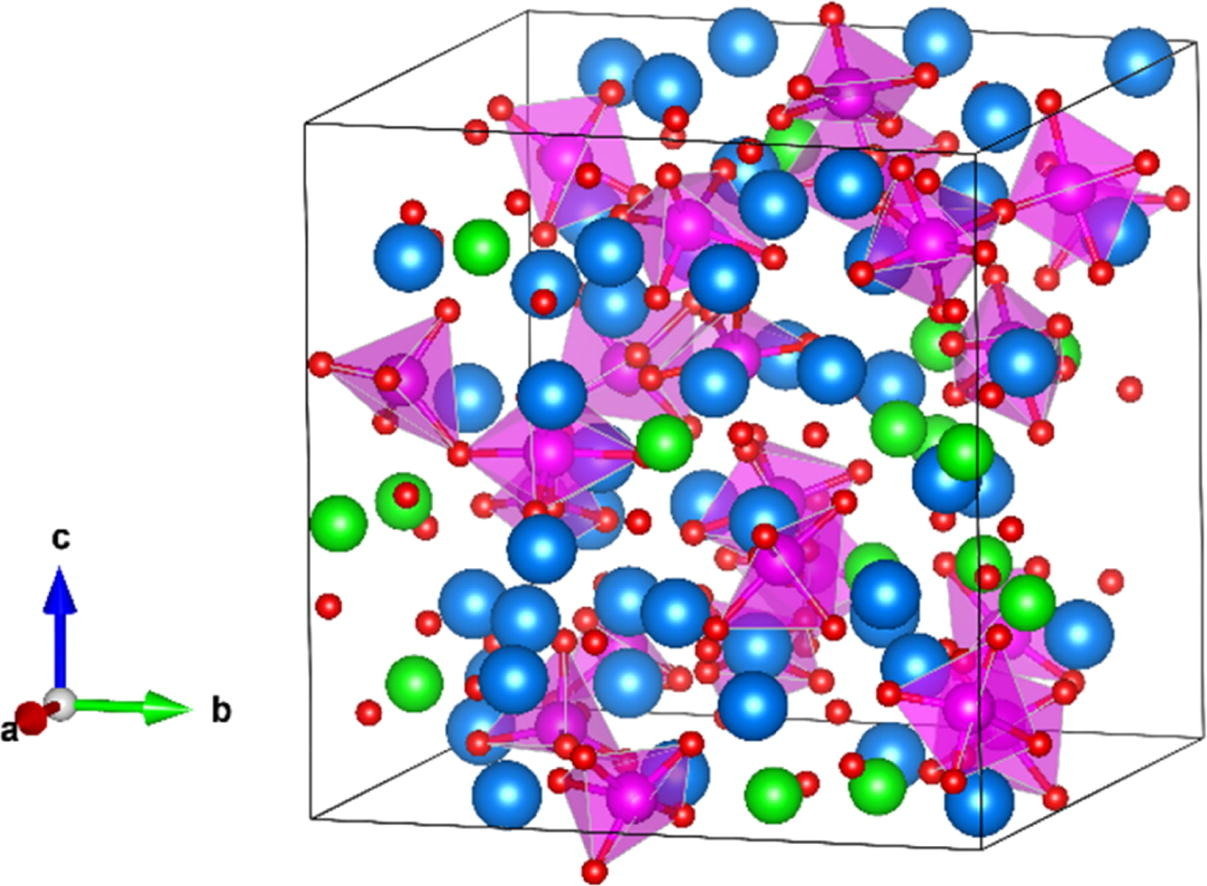
Molten salts
Nuclear Reactor by Olena Panasovska from Noun Project
Batteries

Electrolyte by M. Oki Orlando from Noun Project
Charge carriers and electrolytes
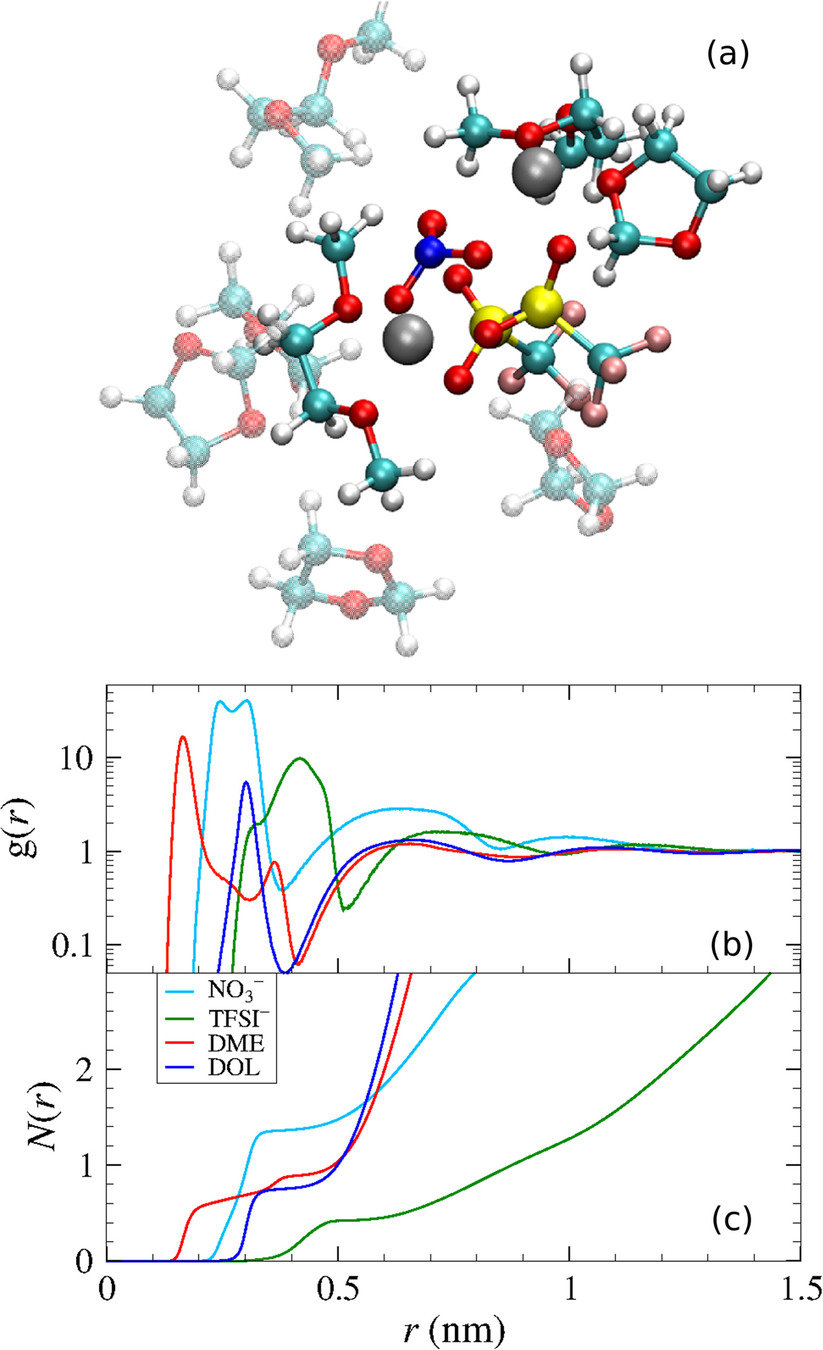
Park, C.; et al. J. Power Sources 2018, 373, 70-78. DOI: 10.1016/j.jpowsour.2017.10.081
Lv, X.; et al. Chem. Phys. Lett. 2018, 706, 237-242. DOI: 10.1016/j.cplett.2018.06.005
Fuel production
Catalysts
Ma, C.; et al. ACS Catal. 2012, 373, 1500-1506. DOI: 10.1021/cs300350b
Complex simulations are hindered by our current force fields
Lv, X.; et al. Chem. Phys. Lett. 2018, 706, 237-242. DOI: 10.1016/j.cplett.2018.06.005
Let's model a molten salt

Pro: Fast
Con: Parameters
Classical potential
Quantum chemistry
Pro: Accurate
Con: Cost
DFT
N^{3-4}
MP2
N^5
CCSD(T)
N^7
Confidence
Confident predictions require explicit molecular simulations
Computational modeling
Cost
AIMD
Classical MD
Implicit/explicit
Implicit
Confidence
Cost

Screening
approach
Promising candidates
Search space
without experimental data
Solvation treatments
Goal
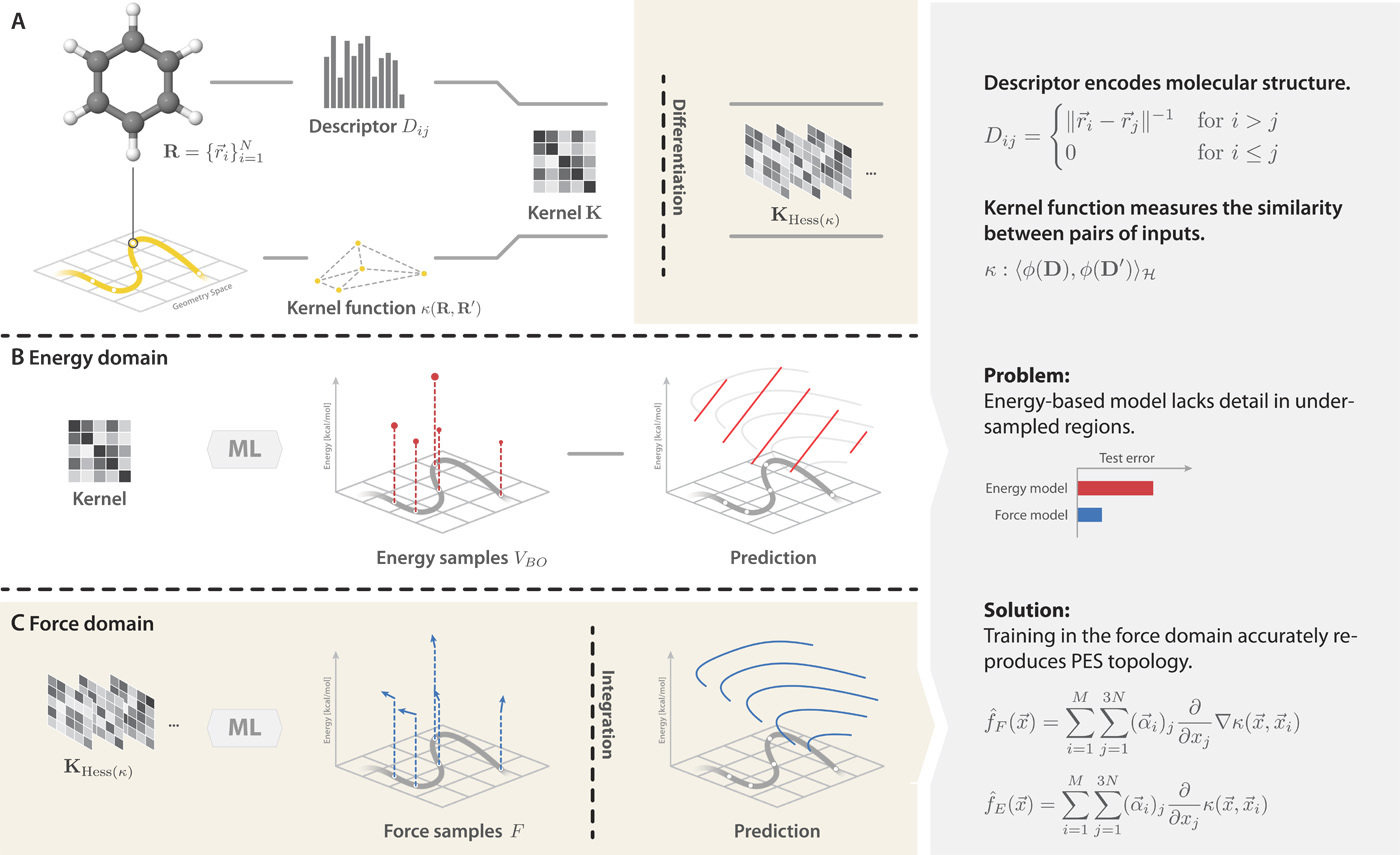
Machine learning potentials accelerate quantum chemical predictions

Structure
ML potential
Energy and forces
Quantum
chemistry
Machine learning potentials accelerate quantum chemical predictions
Most ML force fields use per-atom contributions
Calculate total energies with QC
Training a typical ML potential
Sample tens of thousands of configurations


Approximation: atomic contributions can reproduce total energy
Examples: DeePMD, GAP, SchNet, PhysNet, ANI, . . .
E_{total} \approx \sum\limits^{N_{atoms}} \varepsilon_i
Known
Learned
with a local descriptor
Global descriptors provide superior data efficiency


Local
Global


Encodes each
atom
Encodes entire
structure
Many descriptors and parameters
Single descriptor
Training on force enables better force field interpolation
Training on forces provides more information about the geometry and energy relationship
Chimiela, S. ; et al. Sci. Adv. 2017, 3 (5), e1603015. DOI: 10.1126/sciadv.1603015
Gradient-domain machine learning (GDML)
Better interpolation
Global descriptor
Training on forces
+
requires 1 000 structures instead of 10 000+
=
Global descriptors are not size transferable
System size is still a limiting factor
Global
Fewer structures enables higher levels of theory
Local
No
Tons of sampling
Descriptor
Size transferable?
CCSD(T)
How can we make GDML potentials size transferable?
CCSD(T)
What we want
CCSD(T)

CCSD(T)
What we can afford
Transferability with n-body interactions





-76.31270
-76.31251
-76.31273
-228.96298
-0.00831
-0.00705
-0.00700
-228.93794
(-0.02504)
-228.96031
(-0.00267)




1 body
1+2 body
3-body
+
+
=
+
+
=
Add energy
Remove energy
All energies are in Hartrees
Many-body expansion (MBE) unlocks size transferability for expensive methods
MBE: the total energy of a system is equal to the sum of all n-body interactions

Truncate
E =
\sum E_i^{(1)}
\sum\Delta E_{ij}^{(2)}
\sum \Delta E_{ijk}^{(3)}
+ \cdots
+
+
CCSD(T)
Many-body GDML force fields incorporates more physics
Training a many-body GDML (mbGDML) potential
Sample a thousand
configurations
Calculate n-body energy (+ forces) with QC
Calculate total energies with QC
Known
E_{total} \approx \sum E_i^{(1)} + \sum\Delta E_{ij}^{(2)} + \sum \Delta E_{ijk}^{(3)}
E_{total} \approx \sum\limits^{N_{atoms}} \varepsilon_i
Known
Learned



Reproduce physical n-body energies
Approximation: atomic contributions can reproduce total energy
Sample tens of thousands of configurations
Our innovation: Many-body expansion framework accelerated with GDML
- Less training data
- Use higher levels of theory
- Easy to parallelize

Unique opportunity with GDML accuracy and efficiency
If successful
Case study: Modeling three common solvents
Water (H2O)
Acetonitrile (MeCN)
Methanol (MeOH)
Training set
1 000 structures
(instead of 10 000+)
Sampling
n-body structures from GFN2-xTB simulations
Level of theory
MP2/def2-TZVP
ORCA v4.2.0
Useful ML force fields requires accurate relative energies
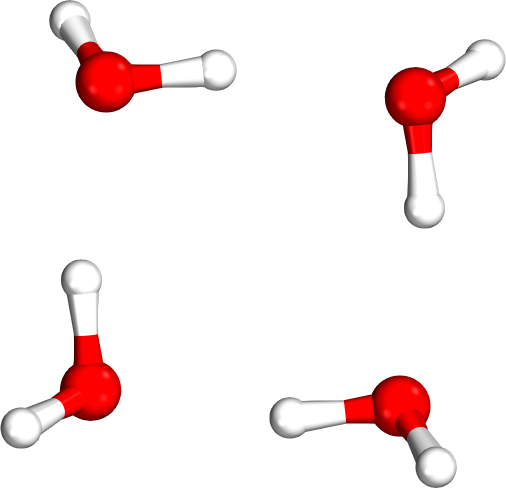
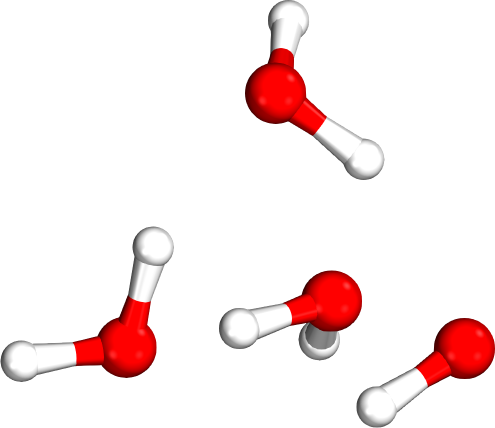
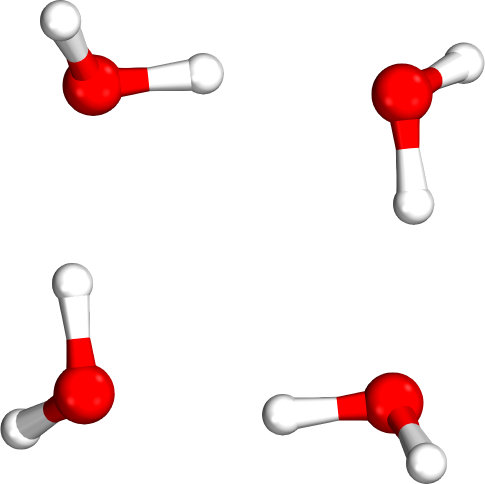
Which tetramer (4mer) has the lowest energy (i.e., global minimum)?
Isomer #1
Isomer #2
Isomer #3
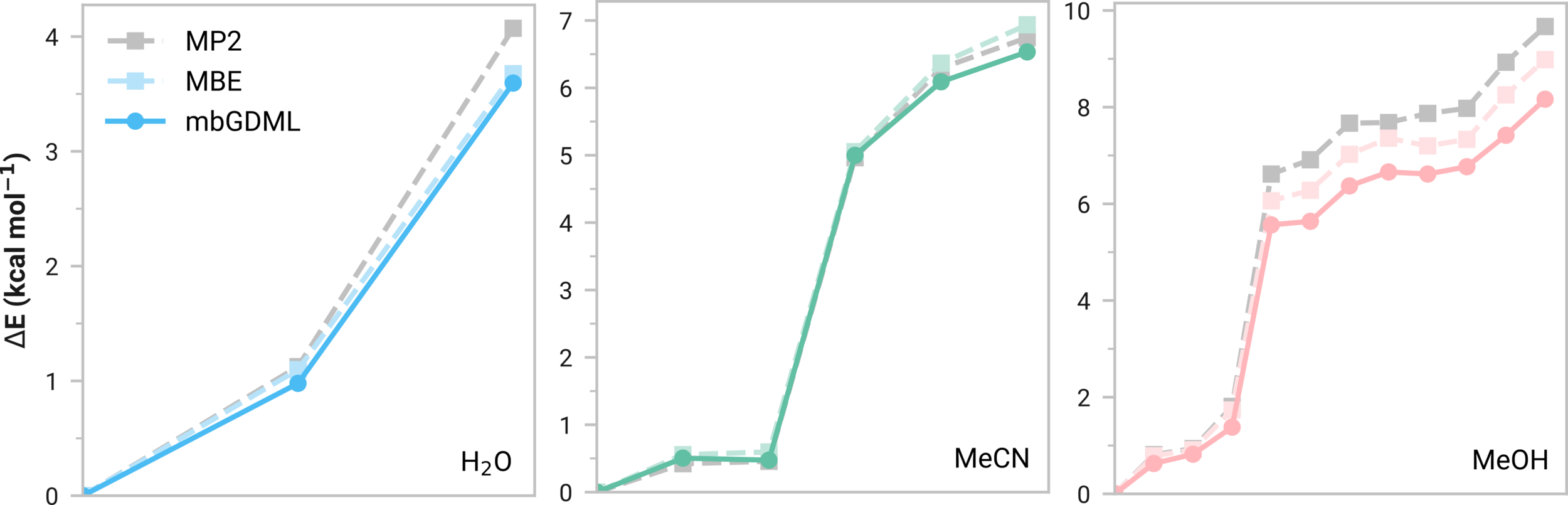
Many-body GDML accurately captures relative energies in tetramers
| System | Energy Error [kcal/mol] | Force RMSE [kcal/(mol A)] |
|---|---|---|
| (H2O)16 | 4.01 (0.25) | 1.12 (0.02) |
| (MeCN)16 | 0.28 (0.02) | 0.35 (0.004) |
| (MeOH)16 | 5.56 (0.35) | 1.79 (0.02) |
mbGDML maintains accuracy on larger systems
Consistent normalized errors
Size transferable
Can we model liquids accurately?
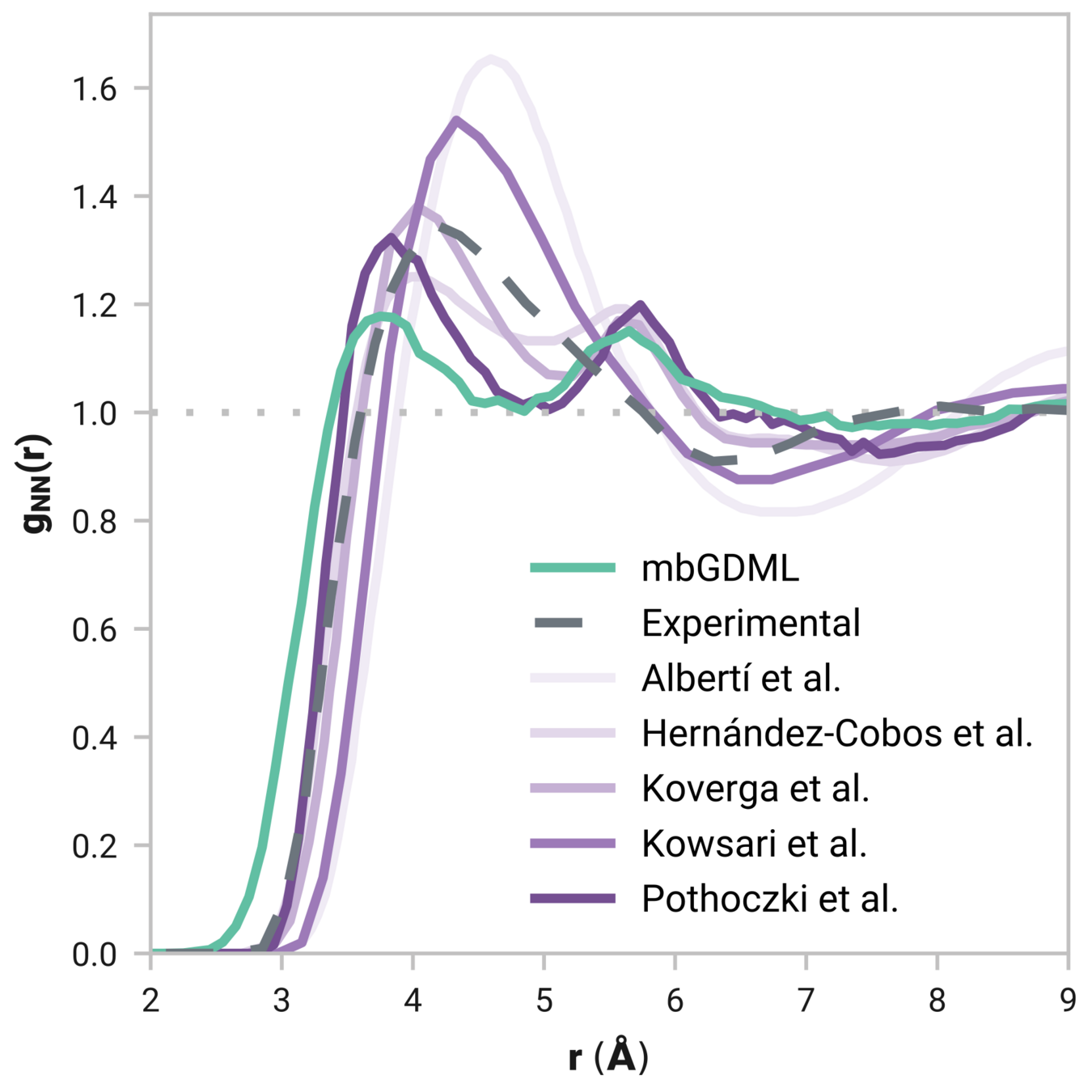
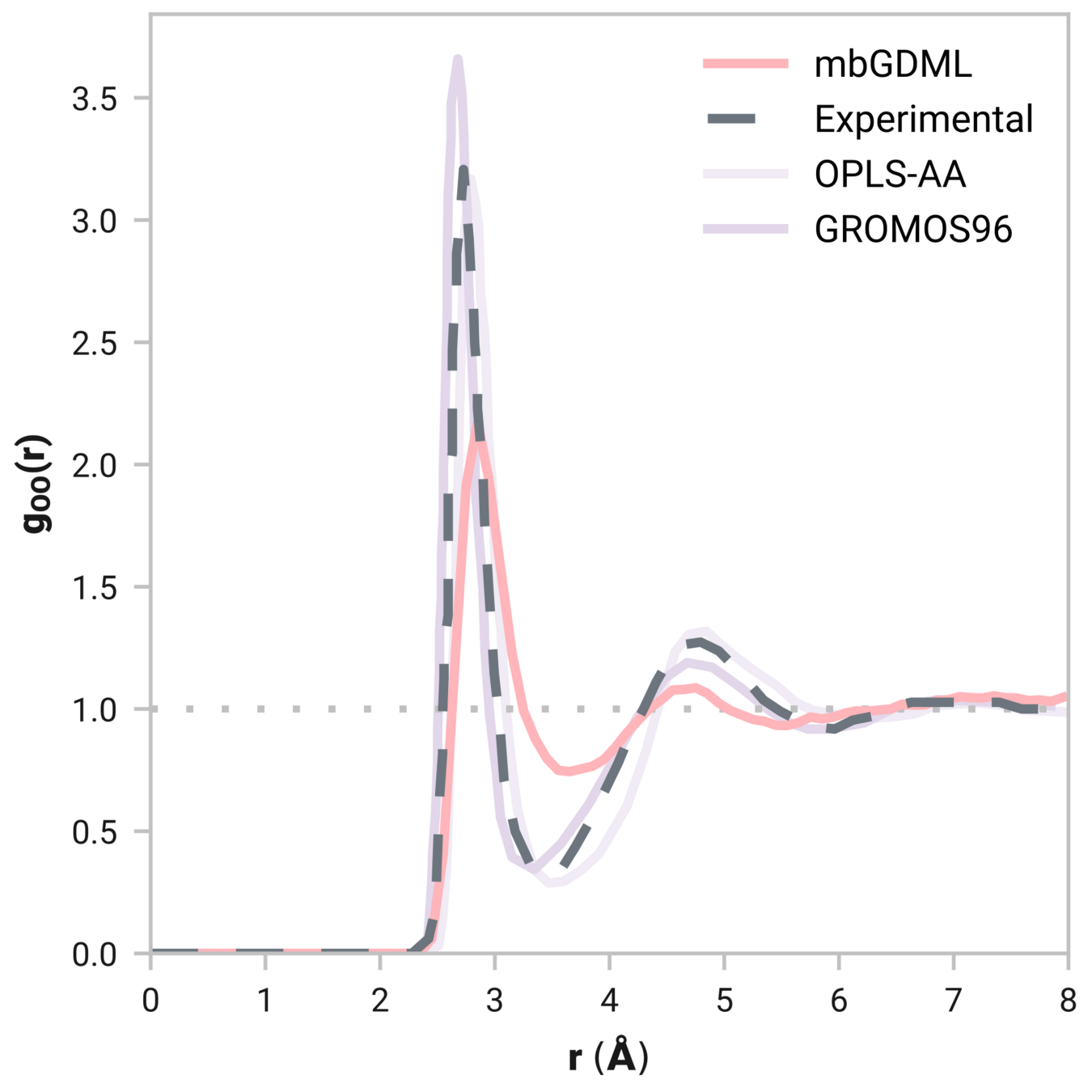
Radial distribution function (rdf)
What we want
r
g(r)

Tells us if we are getting the correct liquid structure
Many-body GDML accurately captures liquid structure
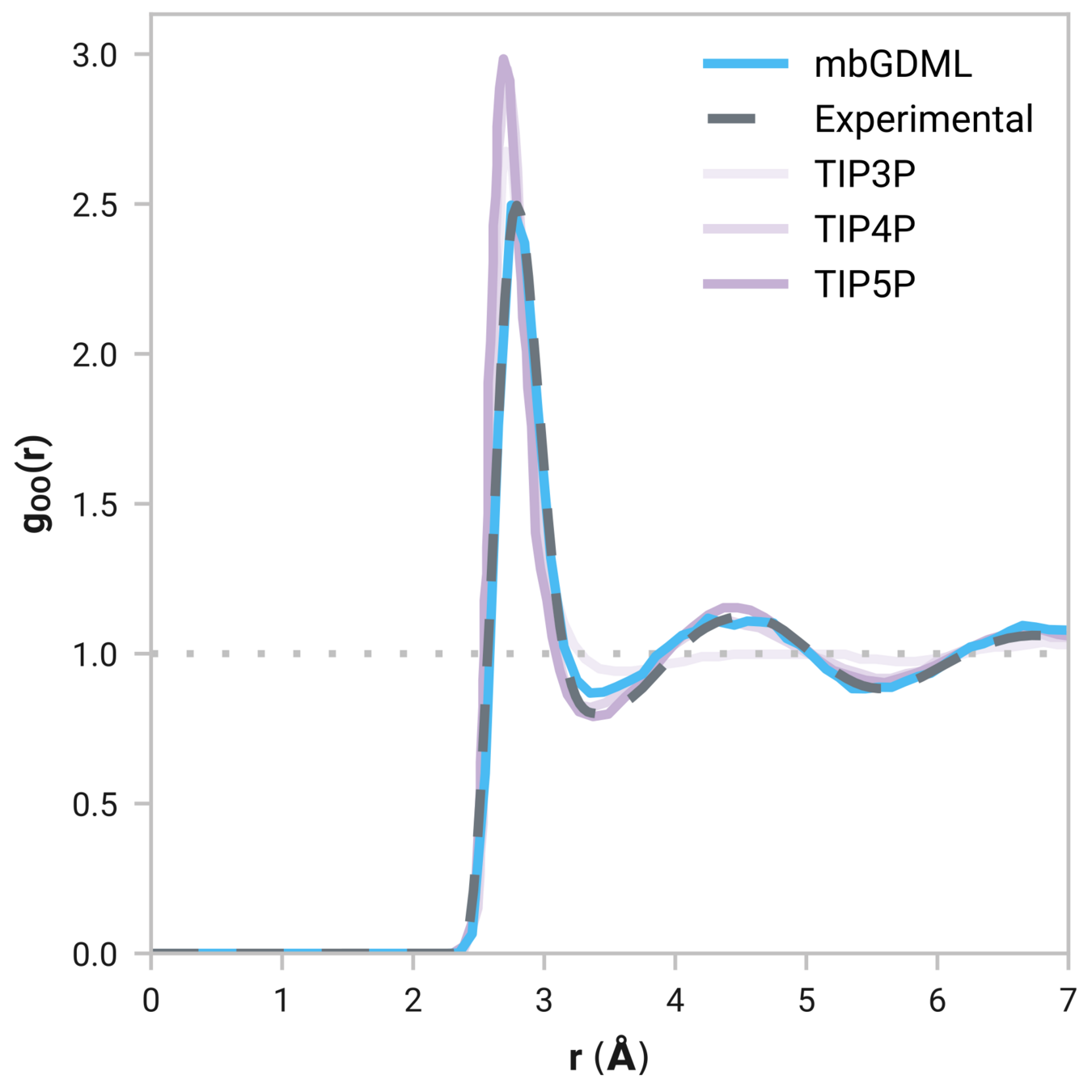
137 H2O molecules
67 MeCN molecules
61 MeOH molecules
Reminder: We have only trained on clusters with up to three molecules
Time for 10 ps MeCN simulation:
mbGDML 19 hours
\approx
MP2 23 762 years
\approx
20 ps NVT MD simulations; 1 fs time step; Berendsen thermostat at 298 K
Explicit solvent modeling without experimental data
Classical
Ab initio
ML
mbGDML
Training

Speed
Accuracy


Scaling


Poor
Excellent
05-storytelling
By aalexmmaldonado
05-storytelling
Presentation for Research Day in the Chemical and Petroleum Engineering Department at the University of Pittsburgh.




