Computational Biology
(BIOSC 1540)
Aug 29, 2024

Lecture 02:
DNA sequencing
Announcements
- Assignment 01 will be published tonight or tomorrow.
- What material will you be responsible for
- Anything covered on slides
- Anything under the "Readings" subsection on the lecture page
- TopHat question about project.
After today, you should be able to
1. Construct a general workflow intrinsic to DNA sequencing experiments.
2. Delineate the core principles underlying Sanger sequencing.
3. Conduct a comparative analysis of Illumina sequencing vis-à-vis Sanger sequencing.
4. Explicate the fundamental principles governing Nanopore sequencing technology.
How do we acquire our DNA sample?
Computationalists still need to understand the underlying source of our data
Let's start with a bacterial culture

Fun fact: Pitt has a beer brewing class (ENGR 1933)
We let our bacterial culture produce our products of interest

Biotechnology frequently uses massive E. coli cultures to produce biologics
Separate cells from media
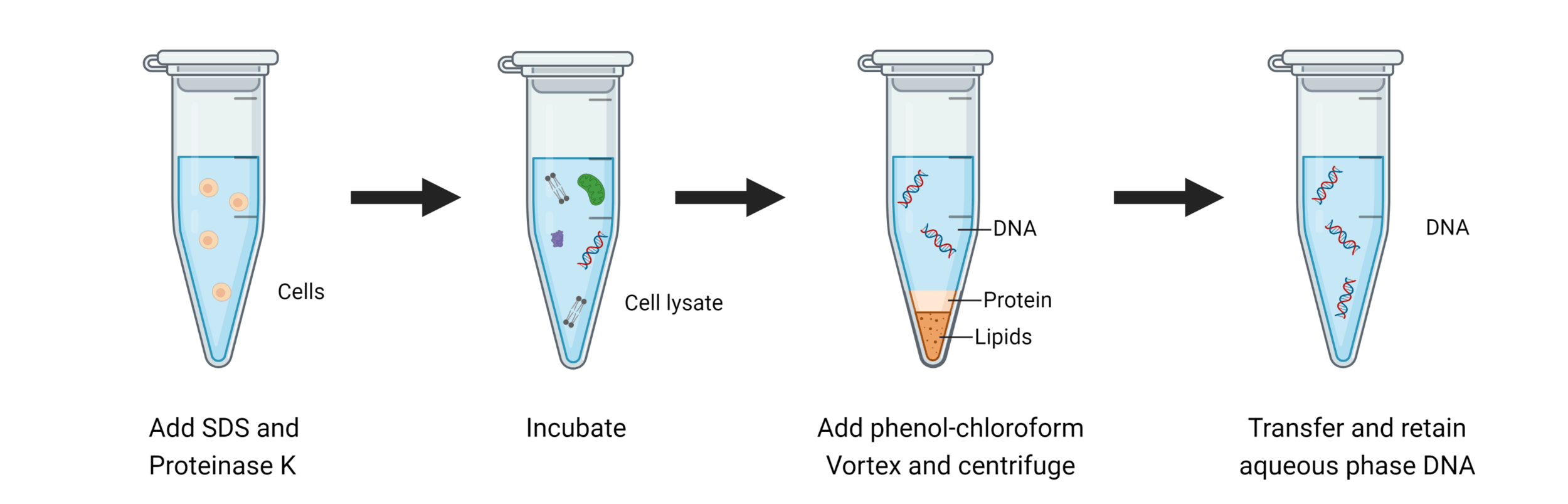
Great! We have our cells, but how can we get DNA out of our cells?
We break open our cells by lysing them
The first step is always to centrifuge and separate our cells and media
Keep the part that has our component of interest (DNA)
How can we lyse cells?
Chemical lysis destabilizes the lipid bilayer and denatures proteins
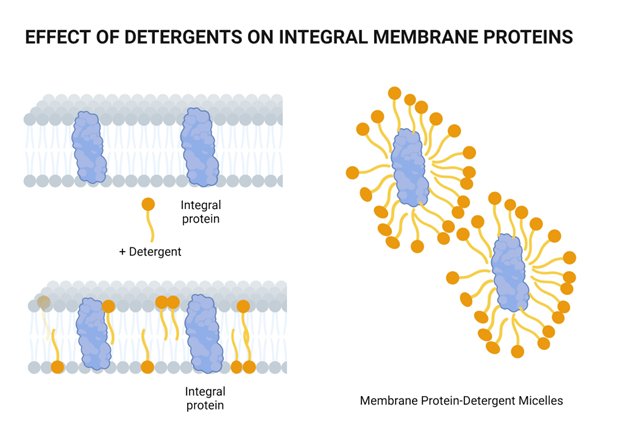
They have a hydrophilic head and hydrophobic tail

Wait, surfactants sound a lot like phospholipids?
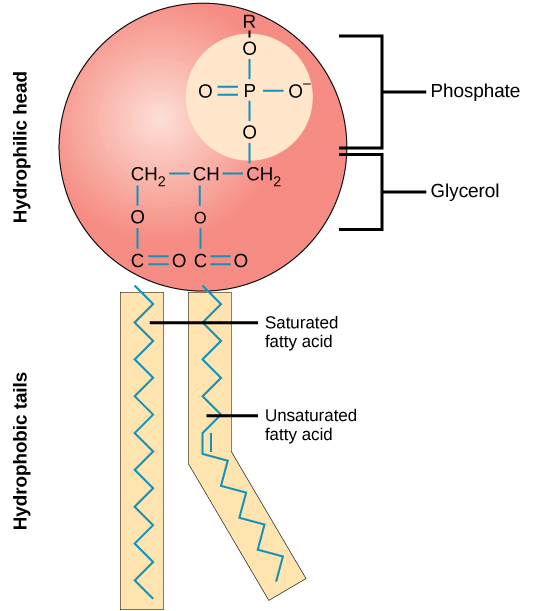
What's the primary difference, and how does this change its behavior?
Surfactants have one hydrophobic tail, which allows them to further penetrate molecular structures
(There are also other methods like sonication.)
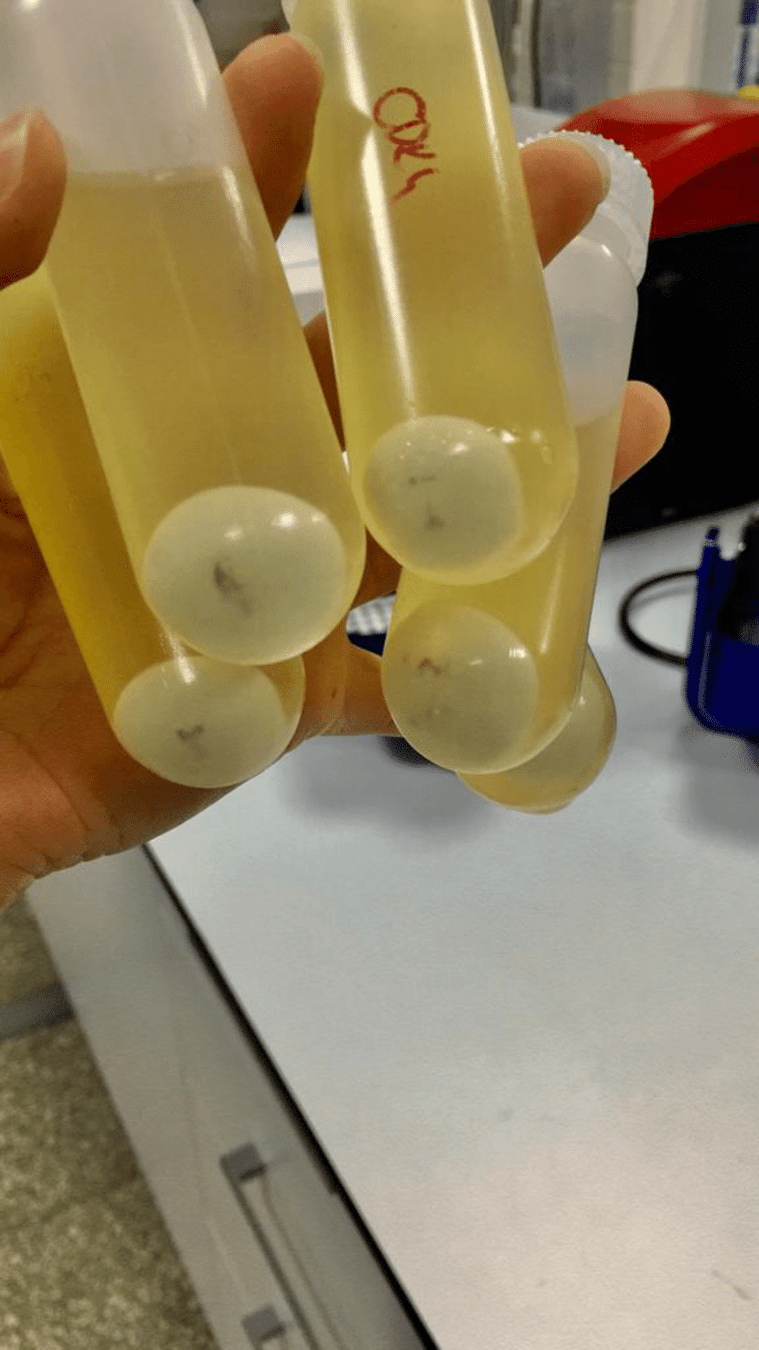
We need to isolate and purify our DNA
Phenol-chloroform extraction uses liquid-liquid separation
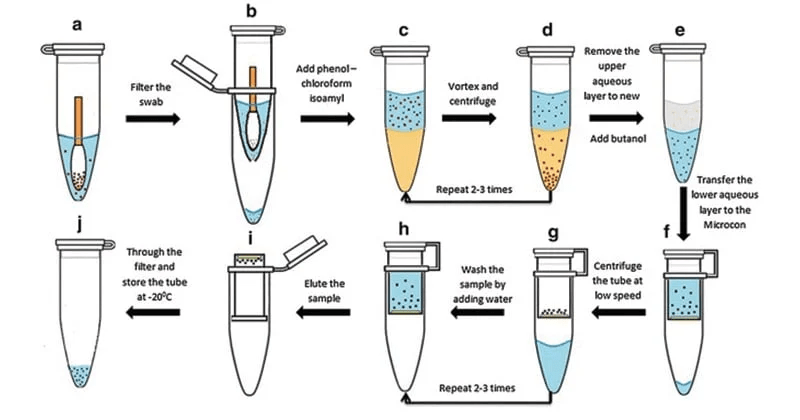
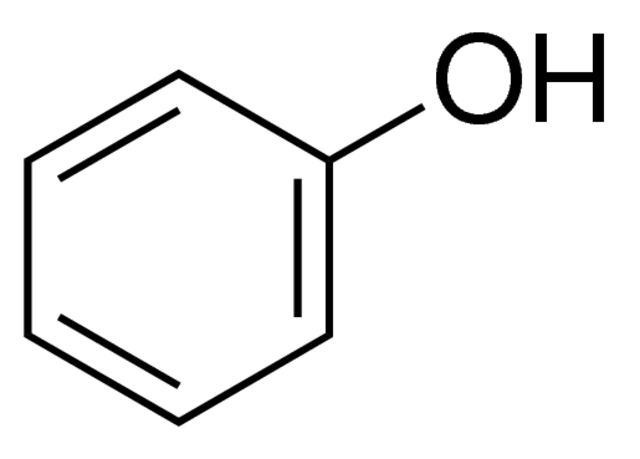
Phenol
Chloroform
Where is our DNA, and why? Which region should we keep?
Aqueous + DNA + RNA
Protein
Lipids + Large molecules
Most labs use highly effective kits

Sample absorbance at 260 nm is correlated to DNA concentration

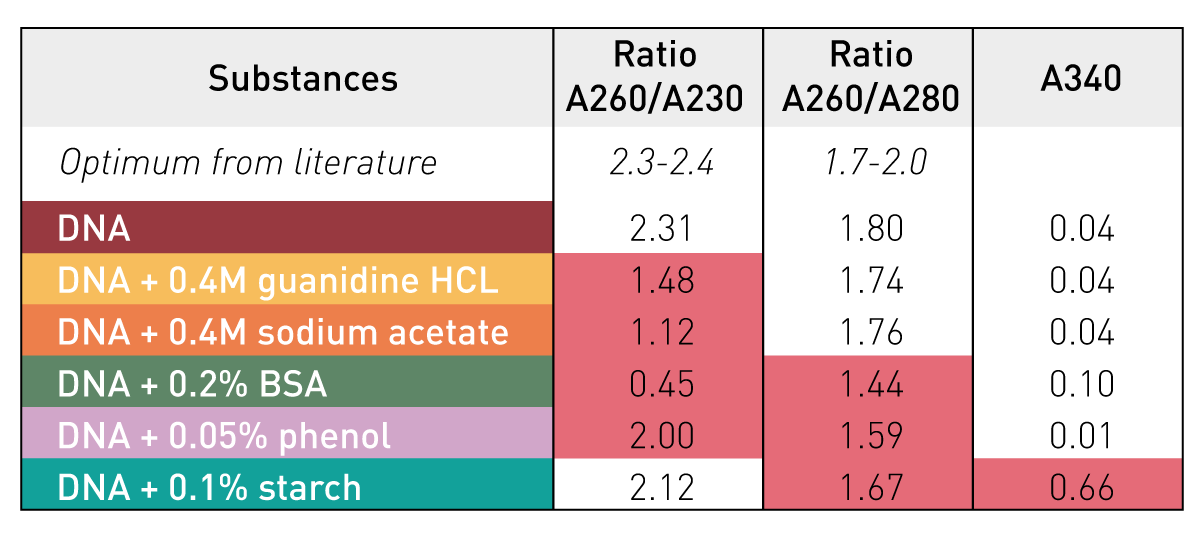
There are some other steps, but let's now assume we have a purified DNA sample at this point
After today, you should be able to
1. Construct a general workflow intrinsic to DNA sequencing experiments.
2. Delineate the core principles underlying Sanger sequencing.
3. Conduct a comparative analysis of Illumina sequencing vis-à-vis Sanger sequencing.
4. Explicate the fundamental principles governing Nanopore sequencing technology.
Our main problem: Determine the precise ordering of nucleotides
DNA elongation happens rapidly and continuously
We use DNA polymerase + excess nucleotides to make copies of DNA
3' OH is required for DNA elongation

What happens if we don't have the 3' OH?
We cannot add another nucleotide
Di-deoxynucleotides stop replication
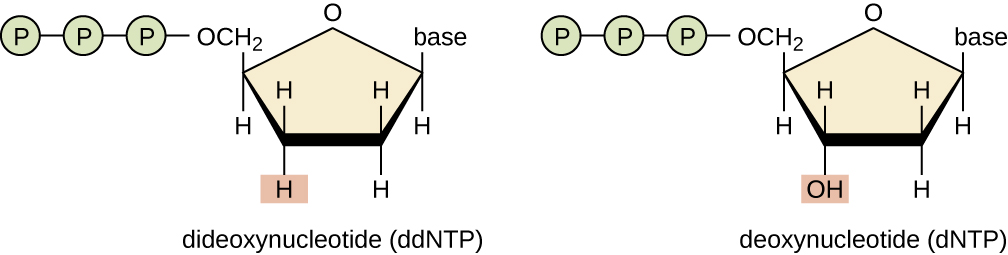

ddNTP will randomly stop DNA elongation
We will be left with DNA strands of variable length
When DNA polymerase adds a
ddNTP
, it cannot add any other
nucleotide
Ratio is usually
1
:
100
By sorting DNA fragments by length, we can see what the last nucleotide is

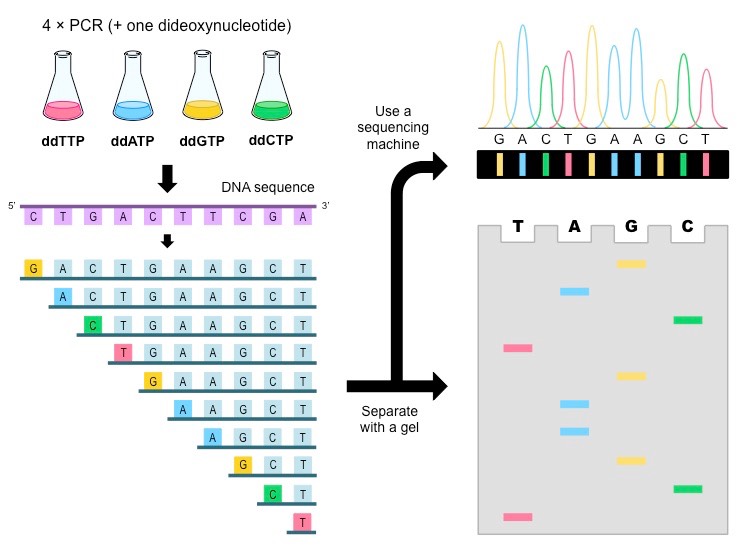
Original setup
- Split DNA sample into four beakers
- Add all four dNTPs to each beaker
- Add some amount of radioactive ddNTP in a single beaker
- Add Taq polymerase and let PCR run
Why would we need separate beakers?
Once we have fragments, how can we separate them by length?
Gel electrophoresis!
Cannot differentiate between radioactive nucleotides
We can build our sequence based on what (radioactive) ddNTP is at that position
Now we use fluorescence to distinguish ddNTPs

Only need one PCR!
We also can automate fragment separation
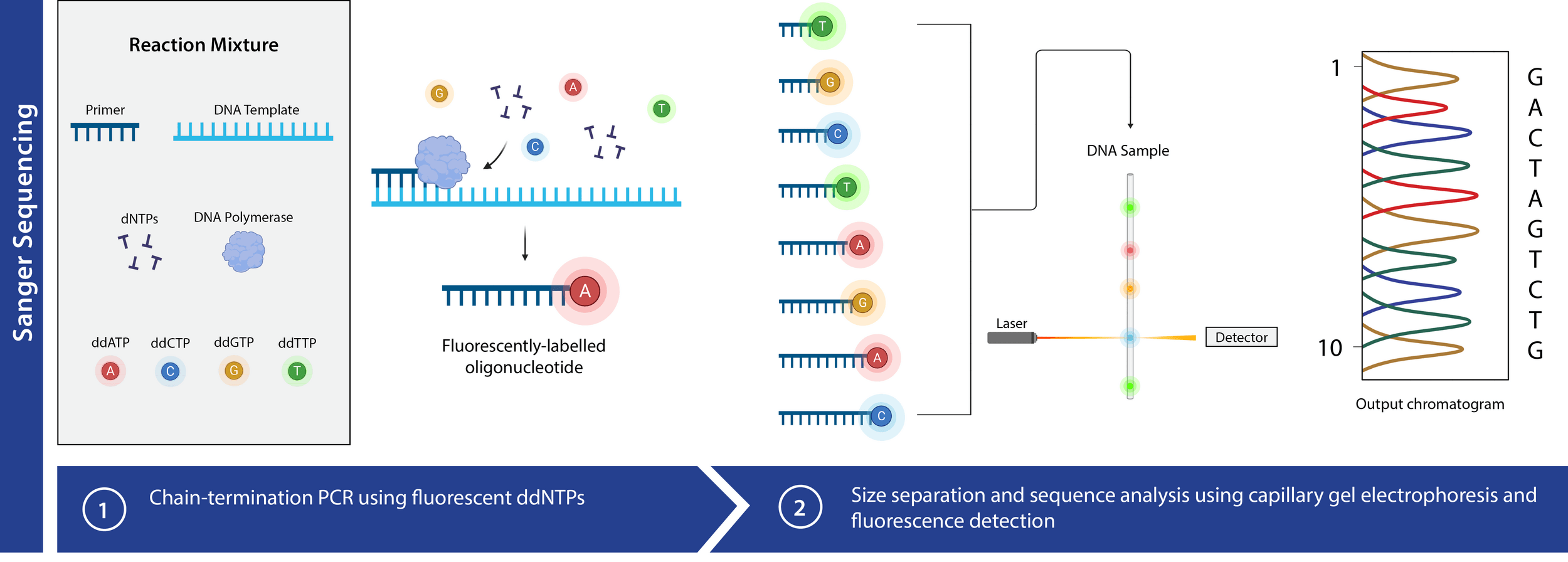
Capillary gel electrophoresis can accelerate fragment length sorting and detection
Unique fluorescence signal per ddNTP produces a chromatogram
Ideal chromatogram

Significant noise up to ~20 basepairs in

Unreliable transport properties
Dye blobs occur from unused ddNTPs

We have fewer longer fragments so signal is weaker

After today, you should be able to
1. Construct a general workflow intrinsic to DNA sequencing experiments.
2. Delineate the core principles underlying Sanger sequencing.
3. Conduct a comparative analysis of Illumina sequencing vis-à-vis Sanger sequencing.
4. Explicate the fundamental principles governing Nanopore sequencing technology.
What is better than promotional materials?
Adapter ligations attach P5 and P7 oligos to facilitate binding to flow cell (Illumina)

Primers are not complementary, so they do not base pair

We locally amplify bound DNA fragments to get clusters of the same sequence

Bridge amplification creates double-stranded bridges
Clusters will give off a stronger signal compared to a single fragment
Double-stranded clonal bridges are denatured with cleaved reverse strands
We repeatedly
-
Add nucleotide
-
Capture signal
-
Cleave fluorophore


Forward
Reverse
Illumina is high throughput and widely used
After today, you should be able to
1. Construct a general workflow intrinsic to DNA sequencing experiments.
2. Delineate the core principles underlying Sanger sequencing.
3. Conduct a comparative analysis of Illumina sequencing vis-à-vis Sanger sequencing.
4. Explicate the fundamental principles governing Nanopore sequencing technology.
What is better than promotional materials?
Nanopores and polymer membrane respond to electrical perturbations
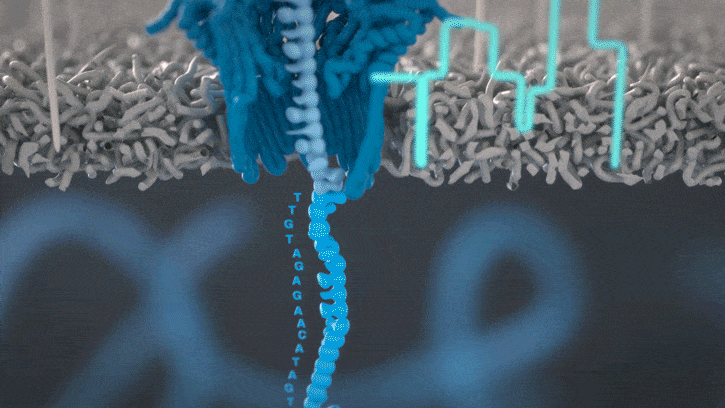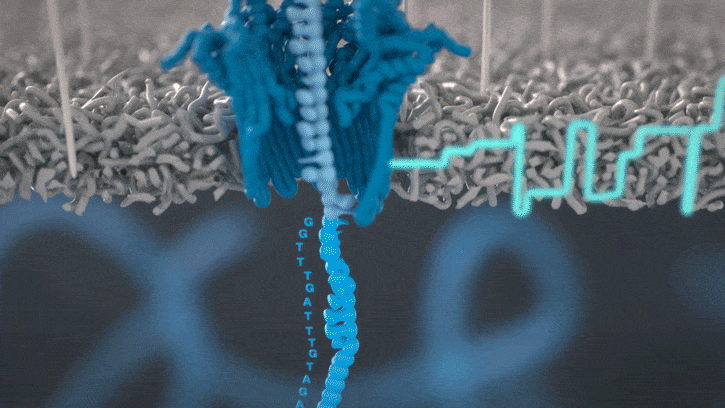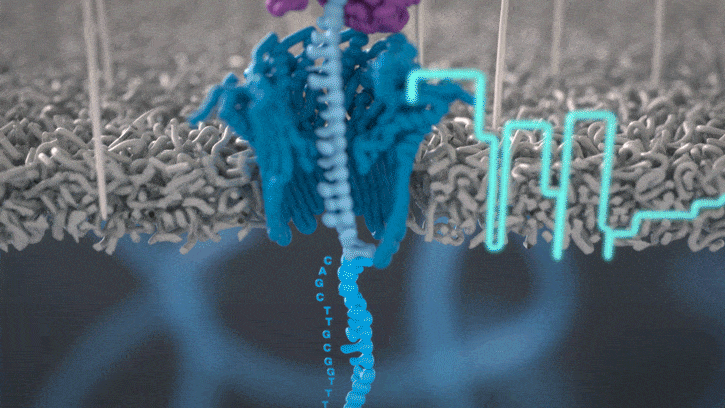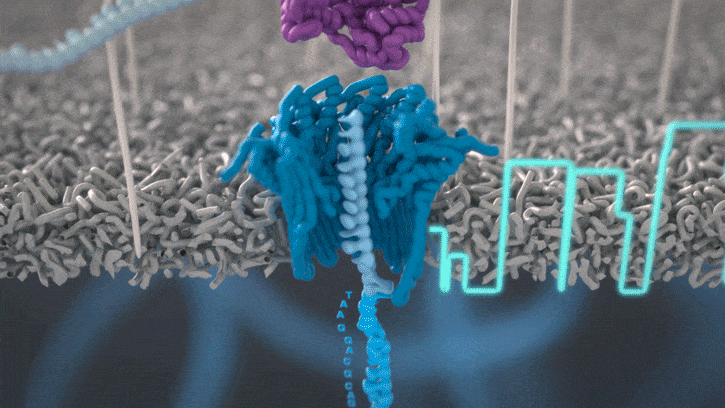

ML algorithms predict and decode sequences

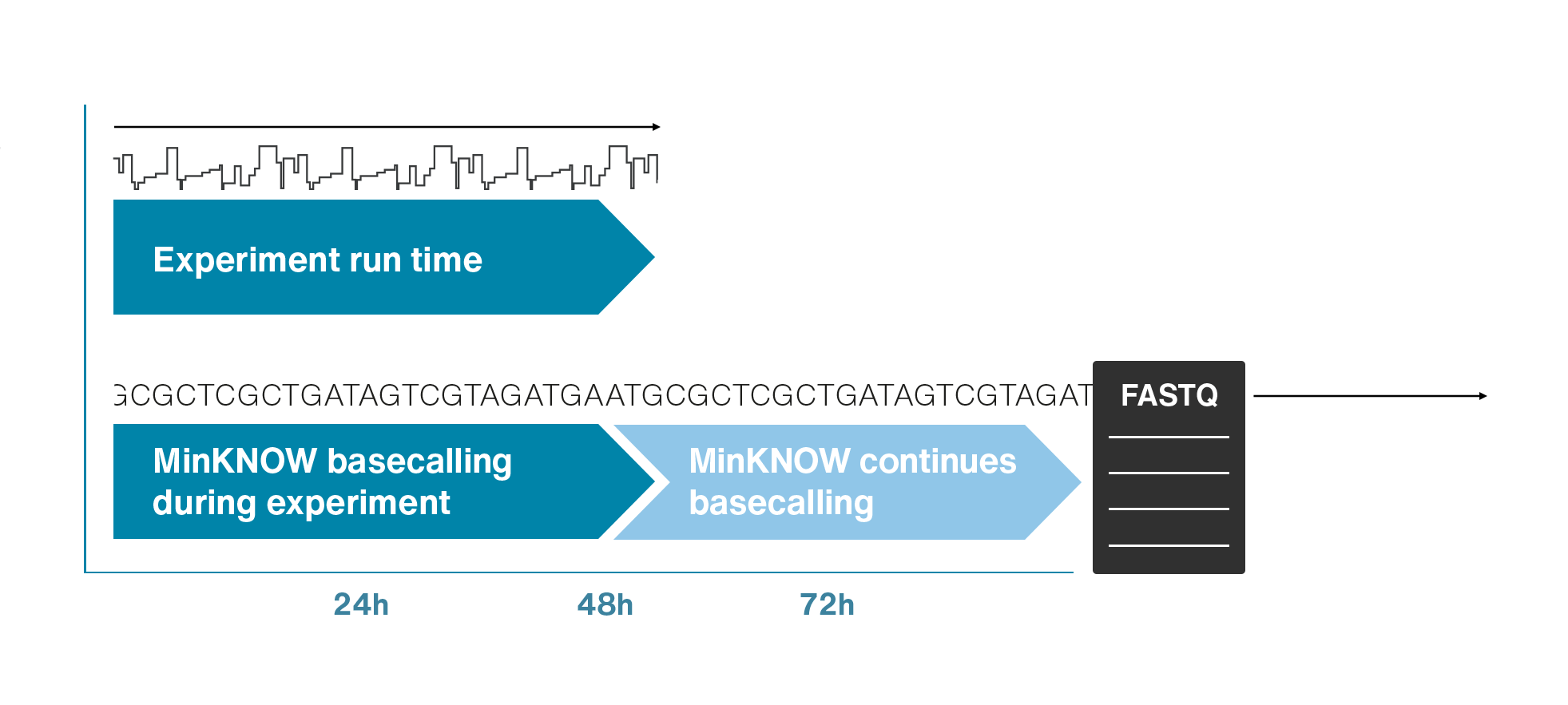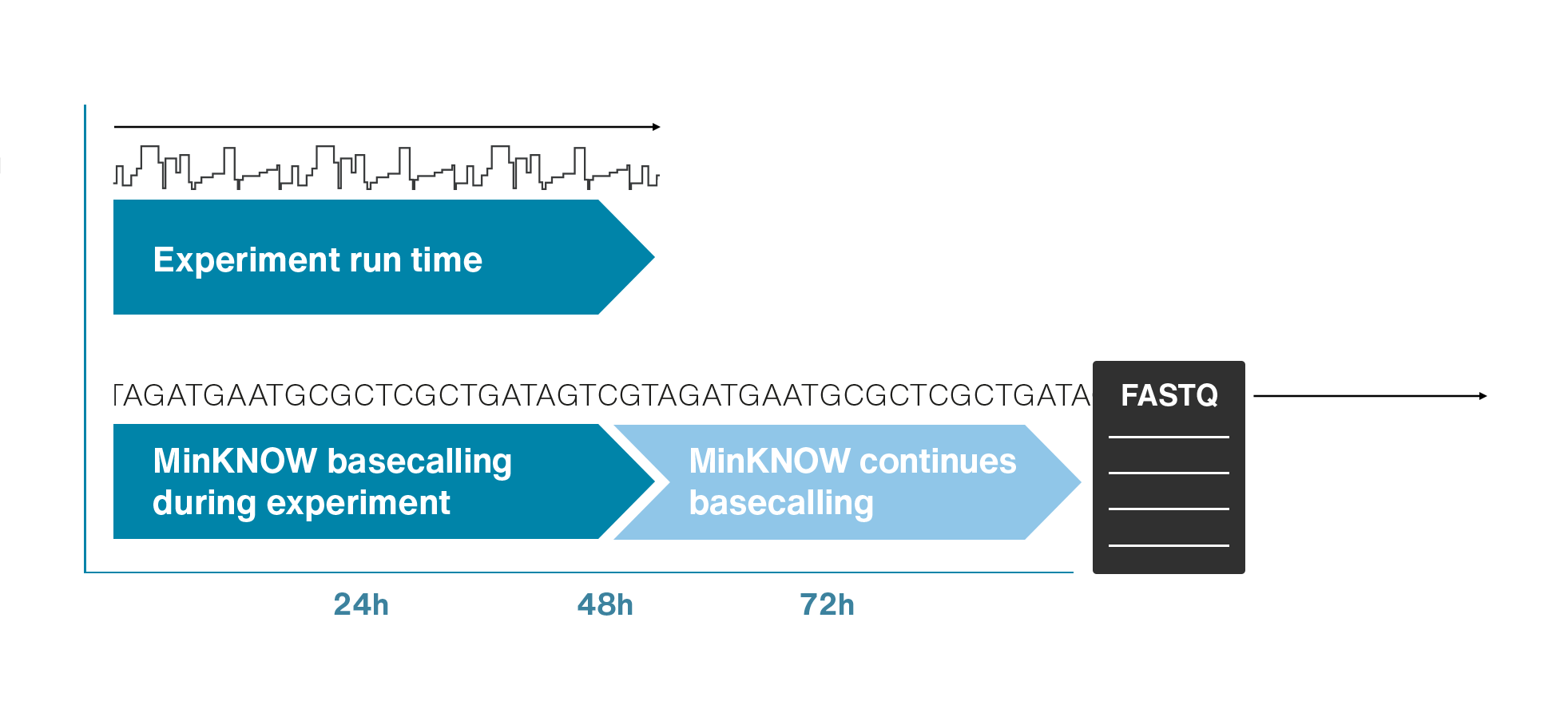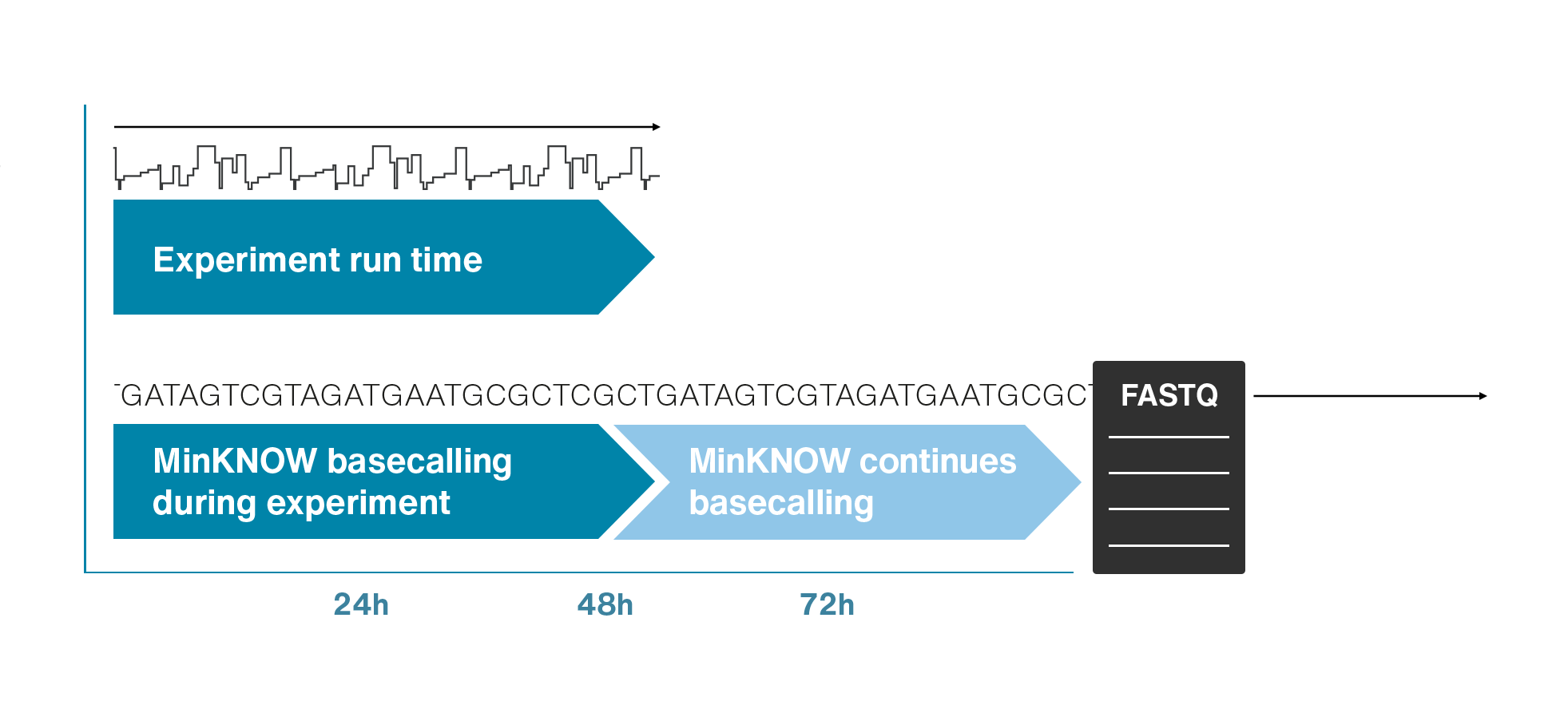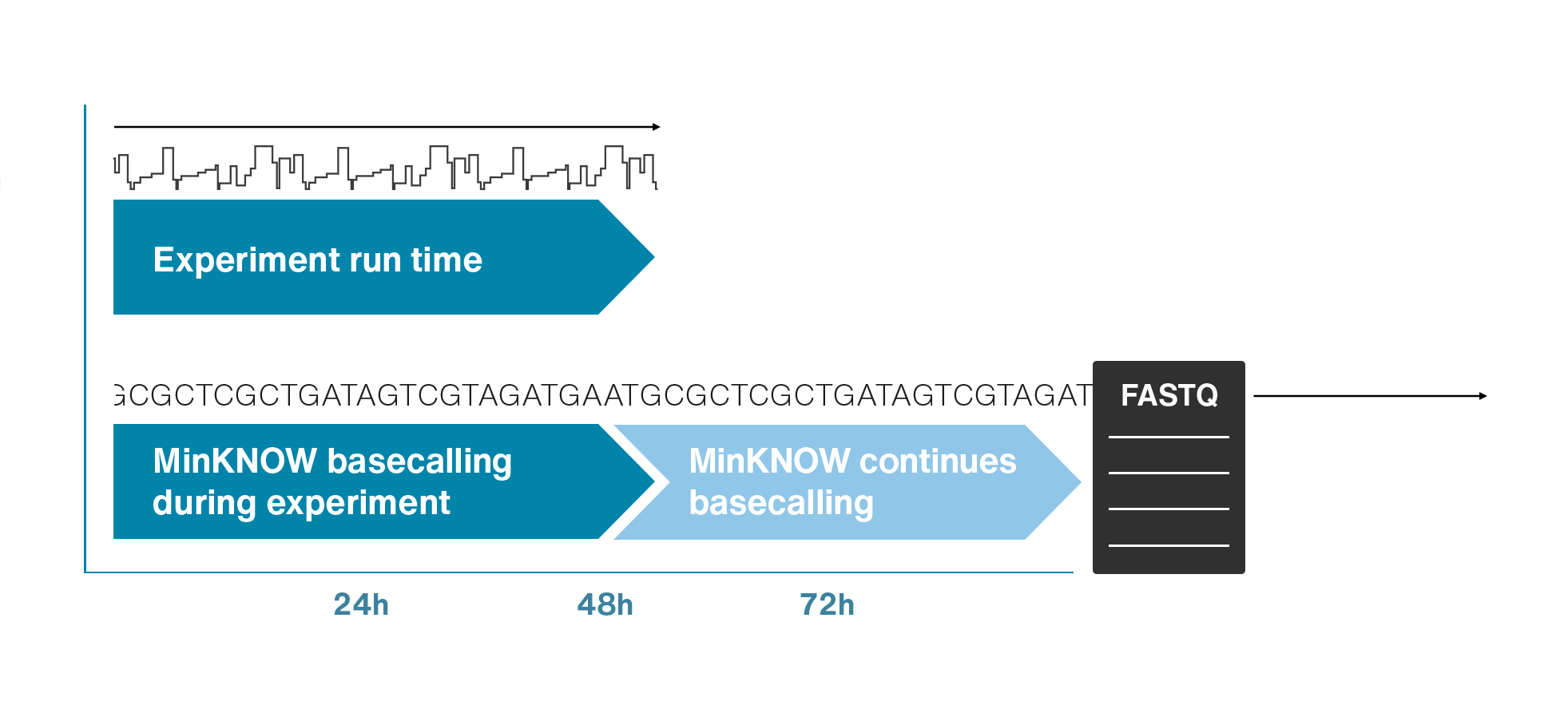
Nanopore gives us much longer reads, which is important for assembling reads into a genome
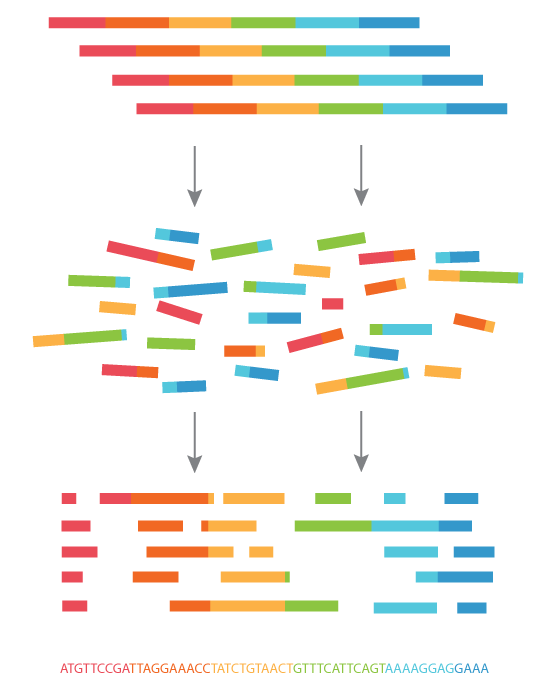
Sneak peek of the next lecture ...
What we sequence
What we want
Use genome assembly!
Before the next class, you should
- Start Assignment 01, which will be released tomorrow.
Lecture 03:
Quality control
Lecture 02:
DNA sequencing
Today
Tuesday
BIOSC 1540: L02 (Sequencing)
By aalexmmaldonado



