Computational Biology
(BIOSC 1540)

Sep 10, 2024
Lecture 05:
Gene annotation
Announcements
After today, you should be able to
1. Explain the graph traversal and contig extraction process in genome assemblers.
2. Understand key output files and quality metrics of genome assembly results.
3. Define gene annotation and describe its key components.
4. Outline the main computational methods used in gene prediction and annotation.
5. Analyze and interpret basic gene annotation data and outputs.
Walking along the graph produces strings
Results in CGTAAAT
CG
GT
TA
AA
AT
AAT
ATG
GGC
GCG
TGG
CGT
GTA
TAA
AAA
AAG
CGA
GAA
AGG
2
2
2
1
1
1
1
1
2
1
1
1
Results in: AATGGCGTAAAGGCGAA
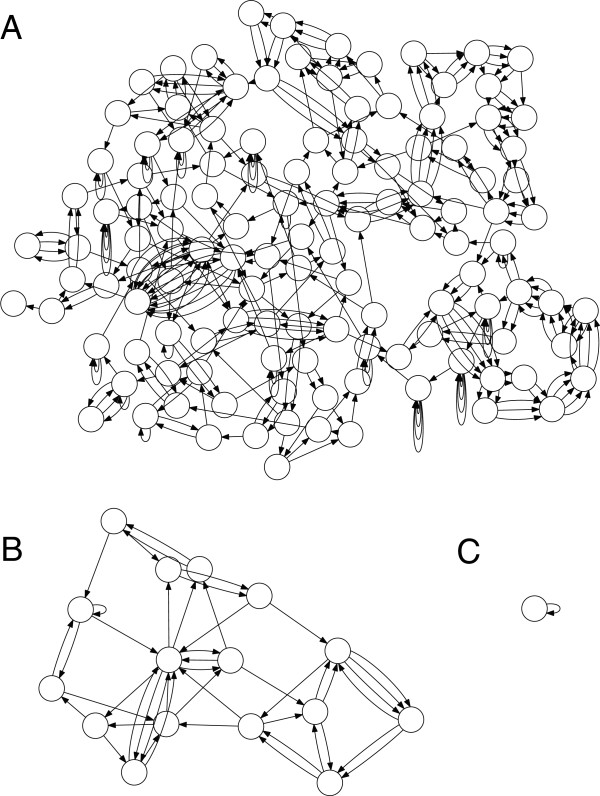
Graphs in practice are not this easy
Graph traversal algorithms are used to extract contigs

General overview
- Select a start node
- Walk along the graph until a dead end or previously visited node is reached
- Backtrack and explore alternative paths
- Repeat for remaining unvisited nodes
Multiple approaches are used and comes down to personal preference
How do we select a starting node?
High coverage: Suggests that the node is likely a true sequence rather than an error
Hubs: Indegree and outdegree != 1

Hubs are shown as filled-in nodes
Hub
Not a hub
Depth-first search explores graph for potential paths (i.e., contigs)
- Start at a chosen vertex (node).
- Mark the current vertex as visited
- Explore an adjacent unvisited vertex
- If no unvisited adjacent vertices exist, backtrack to the last vertex with unvisited adjacent vertices.
- Repeat steps 2-4 until all reachable vertices have been visited.

How do you choose a walk?
How do we choose the "best" path for our contig?
What factors would you look for?
Talk to your neighbors
Long paths are desired but not always reliable due to potential repeats
High, consistent read coverage

Unique, non-branching paths
After today, you should be able to
1. Explain the graph traversal and contig extraction process in genome assemblers.
2. Understand key output files and quality metrics of genome assembly results.
3. Define gene annotation and describe its key components.
4. Outline the main computational methods used in gene prediction and annotation.
5. Analyze and interpret basic gene annotation data and outputs.
Let's get practical with SPAdes

SPAdes is a popular prokaryote genome assembler
Based on De Bruijn graphs with numerous improvements
Error correction with BayesHamming

Build Hamming graphs for k-mers

Undirected edges for Hamming distance of n nucleotide differences
Identify strong k-mers based on clustering (i.e., high similarity)
Estimate read error based on base qualitites
Builds multisized graphs with different k's
Leads to fragmented graphs and helps reduce repeat collapsing
Collapsed, tangled graphs great for low-coverage regions
By using multiple graphs, SPAdes can better handle variable coverage
Large k
Small k
Graph simplification and correction
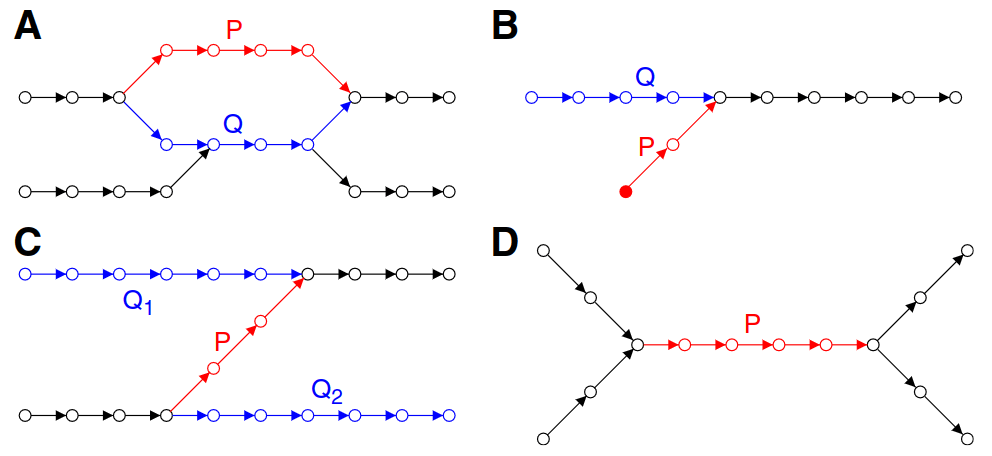
Potential bulge
Removal of a bulge will quickly deteriorate the graph and lose read information
If P needs to be removed, we "project" the information (e.g., coverage) onto Q
P's edges are then removed in the process
Potential tips
Removes P (shortest) and projects information onto Q
Clarification: Paired-ended reads do not always cover our whole insert

Read 1 (forward) and Read 2 (reverse) are stored in FASTQ
If our insert (i.e., DNA sample) is longer than reads, then we don't sequence the inner distance
Should we minimize this inner distance?
False
A gap between paired reads gives us insight into repeated regions
Read 2
ATATATATATATATATAT
Read 1
ATATATATATATATATATAT
Gap
ATATATATATATATATATATATATATATATATATAT
Suppose I have an "AT" repeat for both Read 1 and 2
The assembler will have to figure out if these are overlapped or separated, but by how far?
Having a gap tells me they don't overlap, but for how long?
Knowing length of Read 1, Read 2, and total insert length allows me to calculate gap length
Assembly algorithms (e.g., SPAdes) can estimate this and refine their results
Assemblers provide contigs and scaffolds
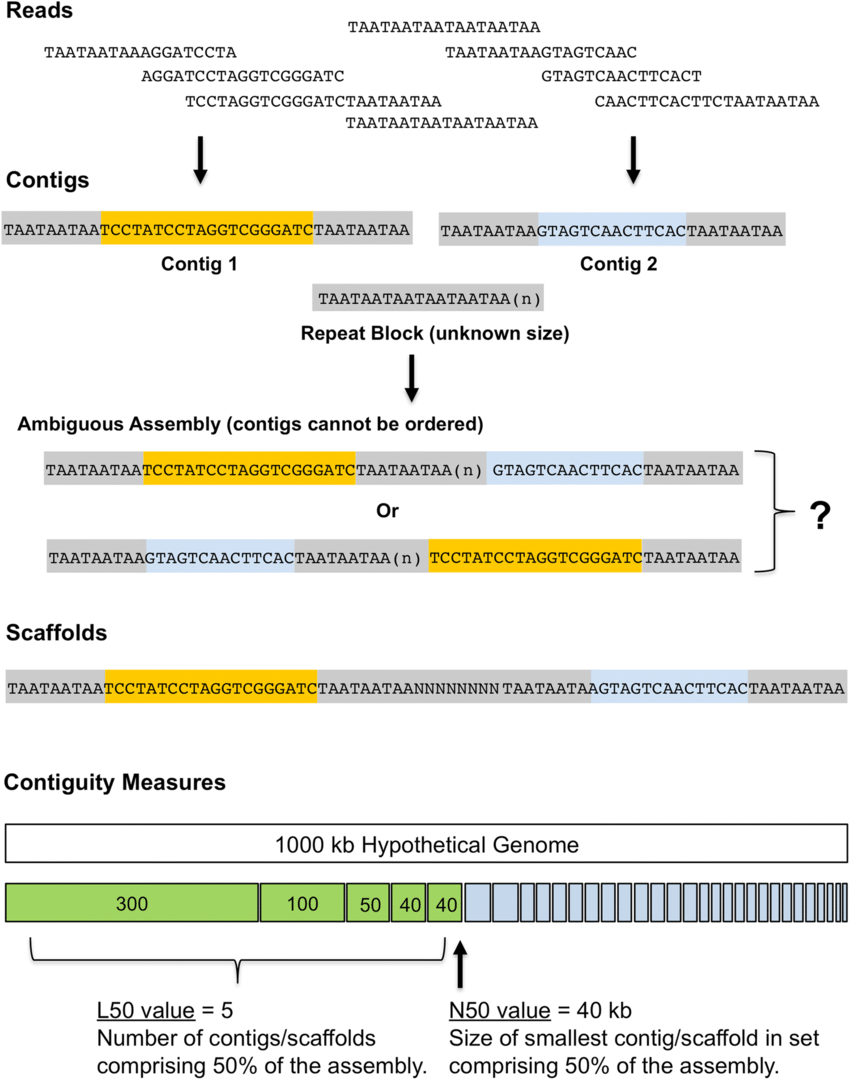

We can visualize this using an assembly graph from a tool called Bandage
Contigs
Scaffolds
Each island contains one or more contigs
Each solid line is called a "node" (Why? I have no idea.) and represent a contig
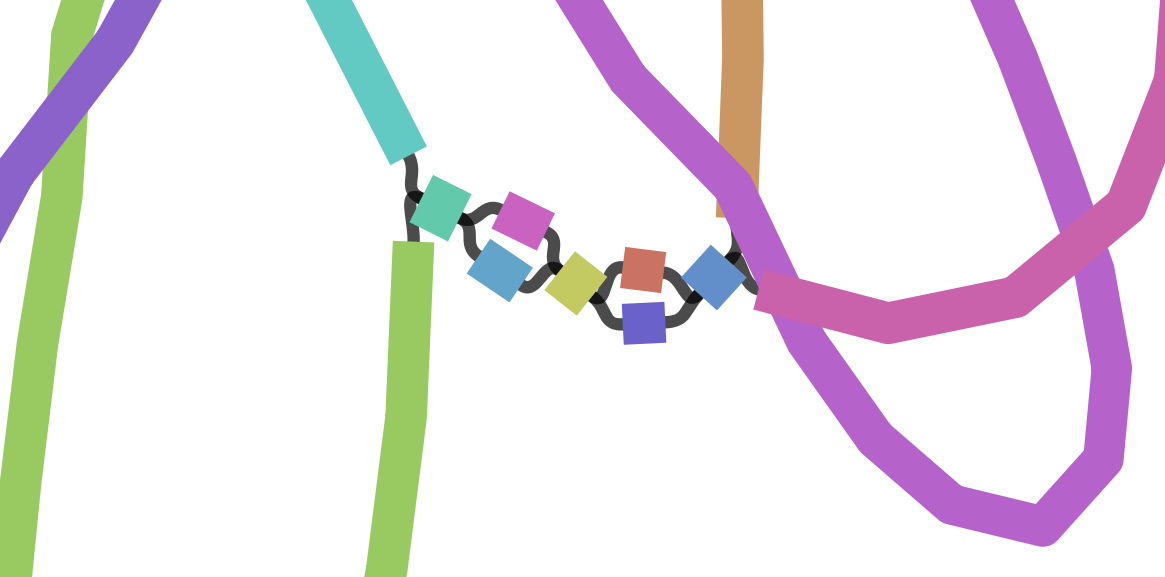
suggests how these contigs connect to form a scaffold
connection
Each
After today, you should be able to
1. Explain the graph traversal and contig extraction process in genome assemblers.
2. Understand key output files and quality metrics of genome assembly results.
3. Define gene annotation and describe its key components.
4. Outline the main computational methods used in gene prediction and annotation.
5. Analyze and interpret basic gene annotation data and outputs.
Annotation is identifying the genetic elements and function in our contigs

Structural annotation identifies critical genetic elements such as genes, promoters, and regulatory elements
Functional annotation predicts the function of genetic elements
Eukaryote annotation is significantly more challenging that prokaryote

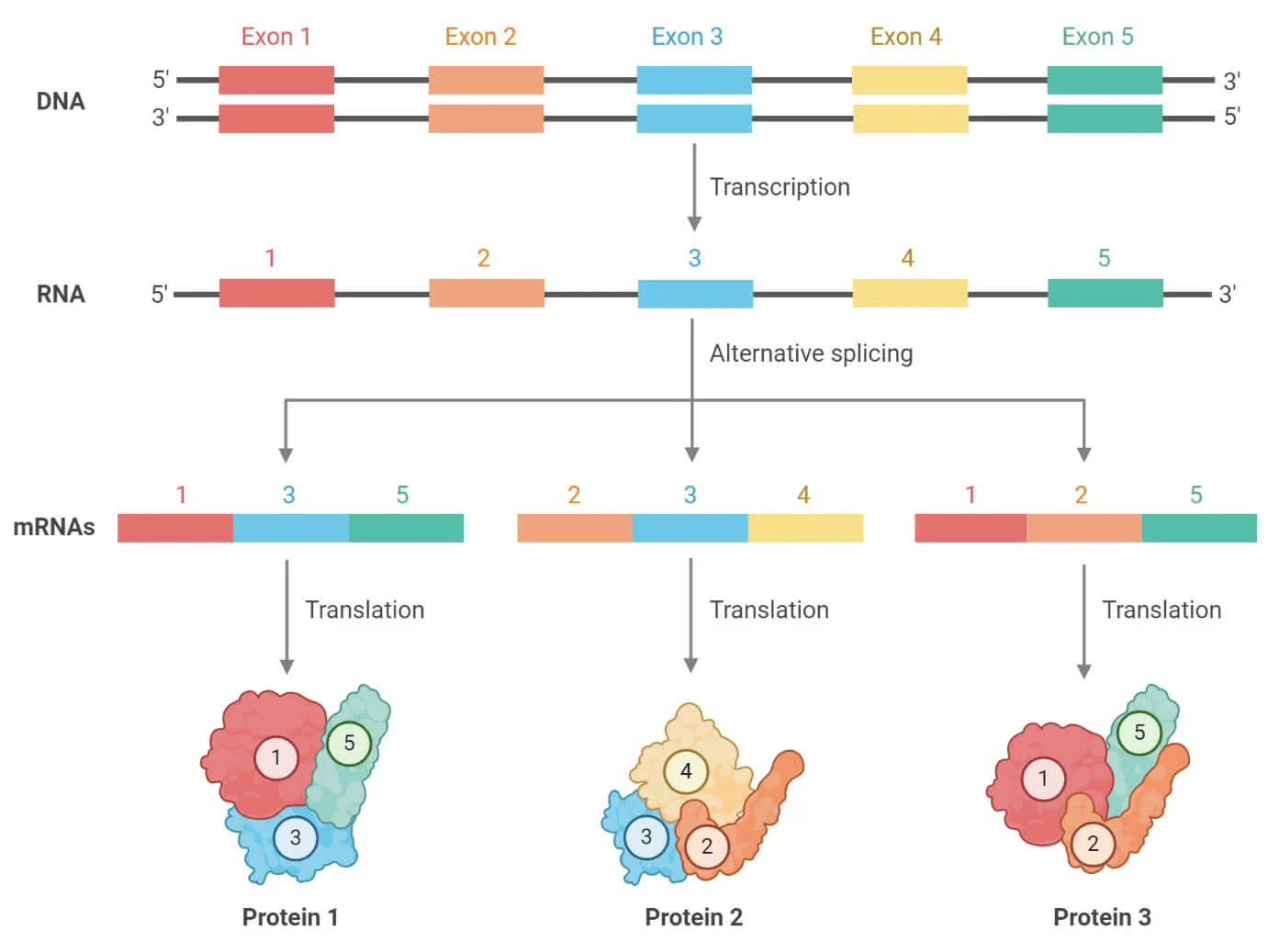
Introns and alternative splicing complicate annotation
Ab initio annotation for prokaryotes is tractable
We will focus on prokaryotes because eukaryotes are way more complicated
Example: Prokka
Example: AUGUSTUS
Prokaryotes
Eukaryotes
Probabilistic models to identify open reading frames

Accuracy demands supporting evidence like mRNA sequencing

After today, you should be able to
1. Explain the graph traversal and contig extraction process in genome assemblers.
2. Understand key output files and quality metrics of genome assembly results.
3. Define gene annotation and describe its key components.
4. Outline the main computational methods used in gene prediction and annotation.
5. Analyze and interpret basic gene annotation data and outputs.
Identify open reading frames (ORF)
(I will use different notation than the paper.)
Seek the standard start codons: ATG, GTG or TTG
Seek stop codons based on the translation table
- TAA, TAG, and TGA for bacteria, archaea, and plant plastids (Code 11)
Score potential ORFs
S_\text{ORF}(n) = 4.25 \left[ S_R(n) + S_T(n) + 0.4 S_U (n) \right] + S_C(n)
Ribosomal binding site motif score
Start type score
Upstream score
Coding score
S_R(n)
S_T(n)
S_U(n)
S_C(n)
RBS score computed from dataset fitting
S_R(n) = \log \left( \frac{R(n)}{B(n)} \right)
Took training data from 12 annotated genomes
Computed frequency of RBS motif bin in
R(n)
- Entire sequence
(Baseline)
- RBS frequency
B(n)
Search for RBS motif after start codon; choose whichever has the lowest bin number

Start
Spacer
RBS
Start codon score given by similar RBS framework
S_T(n) = \log \left( \frac{T(n)}{B(n)} \right)
Took training data from 12 annotated genomes
Computed frequency of start codon in
T(n)
- Entire sequence
(Baseline)
- Start codon frequency
B(n)
Upstream score based on base analysis
Start
Stop
-2 to -1
-44 to -15
By analyzing base frequency in specific upstream regions, their annotation results improved
S_U(n) = w_\text{start} \sum_{i \in P} p_i \left(\text{nuc}_i \right)
Essentially looking for promotors
Coding score computed based on gene enrichment parameters
ATGGCC
CAGCTG
GGGCCC
ACTAGT
Example hexamers called "words"
Computed frequency of nucleotide hexamers called "words" in
G(w)
Compute probability of observing word within the whole genome
within genes
B(w)
Compute the probability of observing word
C(w) = \log \left( \frac{G(w)}{B(w)} \right)
Word coding score
Coding score computed based on gene enrichment parameters
It can be thought of as
"How often does this word appear in genes?"
Gene coding score
S_C (n_\text{start} \ldots n_\text{stop}) = \sum_{i=n_\text{start}}^{n_\text{stop}} C\left( w (i) \right)
Sum hexamer word score and shift over one codon at a time
1
2
3
ATGCATGCTTAG
Results: Sequences that likely encode for proteins
Potential protein
Non-coding
Functional annotation is normally based on protein database search
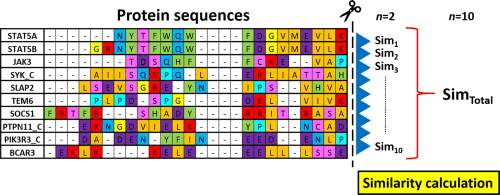
Similarity search will be our topic for Thursday
After today, you should be able to
1. Explain the graph traversal and contig extraction process in genome assemblers.
2. Understand key output files and quality metrics of genome assembly results.
3. Define gene annotation and describe its key components.
4. Outline the main computational methods used in gene prediction and annotation.
5. Analyze and interpret basic gene annotation data and outputs.
Prokka will provide several outputs

>ECNNONJI_02637 Dihydrofolate reductase
MTLSILVAHDLQRVIGFENQLPWHLPNDLKHVKKLSTGHTLVMGRKTFESIGKPLPNRRN
VVLTSDTSFNVEGVDVIHSIEDIYQLPGHVFIFGGQTLFEEMIDKVDDMYITVIEGKFRG
DTFFPPYTFEDWEVASSVEGKLDEKNTIPHTFLHLIRKKBefore the next class, you should
Lecture 06:
Sequence alignment
Lecture 05:
Gene annotation
Today
Thursday
BIOSC 1540: L05 (Gene annotation)
By aalexmmaldonado



