Computational Biology
(BIOSC 1540)

Sep 12, 2024
Lecture 06:
Sequence alignment
Announcements
- A02 is due tonight at 11:59 pm
- A03 will be posted tomorrow
- My goal is to have all grades done by Sunday!
After today, you should be able to
1. Define sequence alignment and explain its importance in bioinformatics.
2. Describe the basic principles of scoring systems in sequence alignment.
3. Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
4. Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
5. Introduce the concept of multiple sequence alignment (MSA), including its importance and challenges.
Biological sequences reveal evolutionary relationships

We are all familiar with the central dogma and how sequences play a large role
HOX genes: A highly conserved gene

Plays a crucial role in embryonic development, particularly in determining the body plan and specifying the anterior-posterior axis
How do we know it's conserved?
By aligning sequences, we can interpret conservation
Infrequent changes (i.e., high similarity) suggest an evolutionarily conserved sequence

Pairwise alignment reveals relationships between biological sequences

Multiple Sequence Alignment (MSA) extends pairwise comparisons
MSA is the process of aligning three or more biological sequences simultaneously

- Identifies conserved regions across multiple species
- Reveals patterns not visible in pairwise comparisons
Aligning sequences can provide more insight than just evolution
Aligning sequences can provide more insight than just conservation
- Functional annotation
- RNA and protein structure
- Disease-associated mutations
- Vaccine design


After today, you should be able to
1. Define sequence alignment and explain its importance in bioinformatics.
2. Describe the basic principles of scoring systems in sequence alignment.
3. Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
4. Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
5. Introduce the concept of multiple sequence alignment (MSA), including its importance and challenges.
Alignment scores guide the selection of meaningful alignments
- Objectivity: Provides a quantitative measure for comparison
- Optimization: Allows algorithms to find the best alignment
- Significance: Helps distinguish real homology from random similarity
Importance of scoring in alignment selection

Alignment elements reflect evolutionary events in sequences
Match: Identical characters in aligned positions
- Represents conserved regions or no change
- Example score: +1
Gap: Dash (-) inserted to improve alignment
- Represents insertions or deletions (indels)
- Example score: -2
ATGCC
|||||
ATGCCATGCC
|| ||
ATACCMismatch: Different characters in aligned positions
- Indicates substitutions or mutations
- Example score: -1
ATGCC
|| ||
AT-CCGap penalties significantly impact alignment outcomes
Linear gap penalty: Fixed cost for each gap
- Example: -2 for each gap, regardless of length
Affine gap penalty: Different costs for opening and extending gaps
- Example: Gap open = -4, Gap extend = -1
ATGCCCTGGCAT
||| ||
ATG-------ATNumber of gaps
Score
Gap score
7
-2
-14
x
=
ATGCCCTGGCAT
||| ||
ATG-------ATFirst gap
-4
Additional gaps
6
+
Score
Gap score
-1
-10
=
x
Gap penalty choices reflect biological assumptions
Affine penalties:
- Better handling of long indels
- More biologically realistic
ATGCCCTGGCAT
||| ||
ATG-------ATImplications of gap penalty types
Linear penalties:
- Simpler to implement
- May over-penalize long gaps
Biological rationale:
- Single mutation event often causes multi-base indel
- Affine penalties better model this biological reality
-14
-10
vs
Advanced scoring methods enhance alignment accuracy
Sophisticated scoring approaches
- Position-specific gap penalties:
- Reduce penalties in variable regions
- Increase penalties in conserved regions
- Residue-specific gap penalties:
- Adjust penalties based on amino acid properties
- Terminal gap penalties:
- Often reduced to allow end gaps in local alignments
Protein alignments require sophisticated scoring systems
- Proteins have 20 amino acids (vs. 4 nucleotides in DNA/RNA)
-
Simple match/mismatch scoring is insufficient because:
- Some amino acid substitutions are more likely than others
- Chemically similar amino acids often substitute without affecting function
- Evolutionary relationships between amino acids are complex
Substitution matrices quantify amino acid replacement probabilities

- The probability that amino acid i mutates into amino acid j for all pairs of amino acids
- Constructed by assembling a large and diverse sample of verified amino acid alignments
- Reflect the true probabilities of mutations occurring through a period of evolution
- Examples: PAM and BLOSUM
After today, you should be able to
1. Define sequence alignment and explain its importance in bioinformatics.
2. Describe the basic principles of scoring systems in sequence alignment.
3. Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
4. Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
5. Introduce the concept of multiple sequence alignment (MSA), including its importance and challenges.
Global alignment compares sequences in their entirety
Global alignment aligns sequences from start to end
- Key characteristics:
- Attempts to align every residue in both sequences
- Introduces gaps as necessary to maintain end-to-end alignment
- Optimizes the overall alignment score for the entire sequences
- Guarantees finding the optimal global alignment between two sequences
- Basic principle: Build a matrix of alignment scores, then trace back to find the best alignment
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
First, enter zero in our first coordinate (0, 0)
We need to fill in each cell by moving from other cells starting from (0, 0)
Each move "uses" a nucleotide from a row, column, or both
1
0
2
3
4
5
0
1
2
3
4
5
-1
Alignment
-2
-3
A
T
T
A
C
A
A
T
T
C
0
A
-
T
-
-
A
T
-
-4
(Disclaimer: these values are not correct for the final matrix.)
Moving right or down uses a gap and you add the penalty to previous score
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
The last cell in our scoring matrix represents our final score of this alignment
-3
-4
-5
-1
-2
-3
-4
-5
-
A
-
A
-
T
-
T
-
C
Alignment score: -5
A
-
T
-
T
-
A
-
C
-
Alignment score: -5
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
Diagonal moves make a pair
-3
-4
-5
-1
-2
-3
-4
-5
1
-2
A
A
If match: +1
If mismatch: -1
T
A
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
To fill in other cells, we need to find the best move (highest score) from
-3
-4
-5
-1
-2
-3
-4
-5
earlier, adjacent cells
Let's figure out
this score
Option 1
A
A
Match (+1)
0 + 1 = 1
Option 2
A
Gap (-1)
-1 + -1 = -2
Option 3
A
Gap (-1)
-1 + -1 = -2
-
-
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
1
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
-3
-4
-5
-1
-2
-3
-4
-5
1
0
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
Option 1
T
A
Mismatch (-1)
-1 + -1 = -2
Option 2
A
Gap (-1)
-2 + -1 = -3
-
Option 3
Gap (-1)
1 + -1 = 0
-
T
Needleman-Wunsch
Let's align two sequences:
ATTAC
AATTC
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
-3
-4
-5
-1
-2
-3
-4
-5
1
0
-1
-2
-3
0
0
1
0
-1
-1
-1
1
2
1
-2
0
0
1
1
-3
-1
-1
0
2
Repeat until we fill the matrix
The last number represents the best possible alignment score
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
Needleman-Wunsch
We get the alignment by tracing back our moves to (0, 0) from our best score
Starting from the bottom left, what is the last move we made to get this score?
This is the last part of our alignment
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
-3
-4
-5
-1
-2
-3
-4
-5
1
0
-1
-2
-3
0
0
1
0
-1
-1
-1
1
2
1
-2
0
0
1
1
-3
-1
-1
0
2
-
C
0 + -1 != 2
C
-
1 + -1 != 2
C
C
1 + 1 = 2
Needleman-Wunsch
Repeat for the next one
This is the second to last part of our alignment
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
-3
-4
-5
-1
-2
-3
-4
-5
1
0
-1
-2
-3
0
0
1
0
-1
-1
-1
1
2
1
-2
0
0
1
1
-3
-1
-1
0
2
-
T
0 + -1 != 1
C
C
A
-
2 + -1 = 1
C
C
A
T
1 + -1 != 1
C
C
There can be multiple optimal alingments
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
-1
-2
A
T
T
A
C
A
A
T
T
C
0
-3
-4
-5
-1
-2
-3
-4
-5
1
0
-1
-2
-3
0
0
1
0
-1
-1
-1
1
2
1
-2
0
0
1
1
-3
-1
-1
0
2
A
A
-
A
T
T
T
T
A
-
C
C
-
A
A
A
T
T
T
T
A
-
C
C
Global alignment is not always useful
Advantages
- Provides a complete picture of sequence similarity
- Ideal for detecting overall conservation patterns
- Useful for phylogenetic analysis of related sequences
Limitations
- May force alignment of unrelated regions in divergent sequences
- Less effective for sequences of very different lengths
- Can be computationally intensive for long sequences
1. Define sequence alignment and explain its importance in bioinformatics.
2. Describe the basic principles of scoring systems in sequence alignment.
3. Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
4. Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
5. Introduce the concept of multiple sequence alignment (MSA), including its importance and challenges.
After today, you should be able to
Local alignment identifies best matching subsequences
Focuses on finding regions of high similarity within sequences
- Does not require aligning entire sequences end-to-end
- Allows for identification of conserved regions or domains
Key characteristics:
- Aligns subsections of sequences
- Ignores poorly matching regions
- Can find multiple areas of similarity in a single comparison
Smith-Waterman
We have a few algorithm changes
Zero is the lowest score (i.e., if negative, make it zero)
Scoring scheme
- Match: +1
- Mismatch: -1
- Gap: -1
| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
0
0
A
T
T
A
C
A
A
T
T
C
0
0
0
0
0
0
0
0
0
1
1
0
0
0
0
2
1
0
0
0
1
3
2
0
1
0
2
2
1
0
0
1
3
0
Start alignment at highest cell
Stop aligning when you encounter a zero
A
T
T
A
T
T
A
T
T
A
T
T
T
T
T
T
Smith-Waterman differs from Needleman-Wunsch in key aspects
Matrix initialization:
- Needleman-Wunsch: The first row and column are filled with gap penalties
- Smith-Waterman: First row and column filled with zeros
Traceback:
- Needleman-Wunsch: Starts from the bottom-right cell
- Smith-Waterman: Starts from highest scoring cell in the matrix
Scoring system:
- Needleman-Wunsch: Allows negative scores
- Smith-Waterman: Negative scores are set to zero
Protein motif identification exemplifies local alignment utility
Can identify functional regions
- Protein domains: Functional or structural units within proteins
- Active sites: Regions directly involved in protein function
- Binding motifs: Short sequences that interact with other molecules
- Signal sequences: Regions that direct protein localization
- Post-translational modification sites: Areas subject to chemical modifications
After today, you should be able to
1. Define sequence alignment and explain its importance in bioinformatics.
2. Describe the basic principles of scoring systems in sequence alignment.
3. Explain the principles and steps of global alignment using the Needleman-Wunsch algorithm.
4. Describe the concept and procedure of local alignment using the Smith-Waterman algorithm.
5. Introduce the concept of multiple sequence alignment, including its importance and challenges.
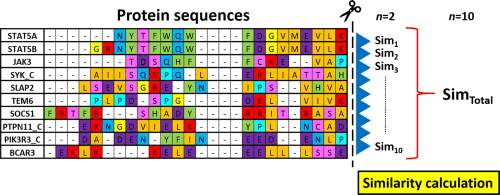
Multiple Sequence Alignment compares three or more sequences simultaneously
Key characteristics:
- Aligns multiple sequences in a single analysis
- Introduces gaps to maximize alignment of similar characters
- Preserves the order of characters in each sequence
Definition of MSA: Arranges three or more biological sequences (DNA, RNA, or protein) to identify regions of similarity
Aims to infer structural, functional, or evolutionary relationships among the sequences
Popular MSA tools include Clustal Omega, MAFFT, and MUSCLE
Before the next class, you should
Lecture 07:
Transcriptomics
Lecture 06:
Sequence alignment
Today
Tuesday
BIOSC 1540: L06 (Alignment)
By aalexmmaldonado



