Computational Biology
(BIOSC 1540)
Oct 10, 2024
Lecture 12:
Protein structure prediction
Announcements
- No class on Tuesday (10/15)
- No office hours (mine or UTA) next week - will resume on 10/22
- Will have Programming+ recitations
- A05 will be posted tomorrow
- David Baker, John Jumper, and Demis Hassabis won the Nobel Prize in Chemistry for "computational protein design" and "protein structure prediction"


After today, you should be able to
Why are we learning about protein structure prediction?
Why predict protein structure?
Protein structure dictates interactions, signaling, and biochemical roles
Experimental methods (X-ray, Cryo-EM) provide high-resolution structures but are resource-intensive and time-consuming

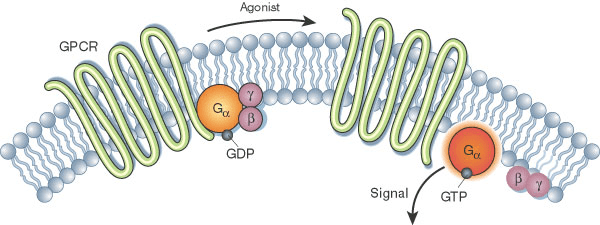

Structural insights can accelerate ... everything?
- Drug Discovery: Designing small-molecule inhibitors or antibodies that target specific protein conformations.
- Biotechnology: Engineering proteins for industrial or therapeutic applications.
- Disease Research: Mutations causing structural defects linked to diseases like Alzheimer’s and cystic fibrosis.
Prediction is critical for the future of biology
Advances in predictive accuracy are opening new frontiers in biology
Structure prediction complements genomics and transcriptomics to create a holistic understanding of biological function
Integrating predictive models with experimental data is the way forward
After today, you should be able to
Identify what makes structure prediction challenging
What makes structure prediction hard: Conformational space
Proteins can adopt a large number of possible conformations
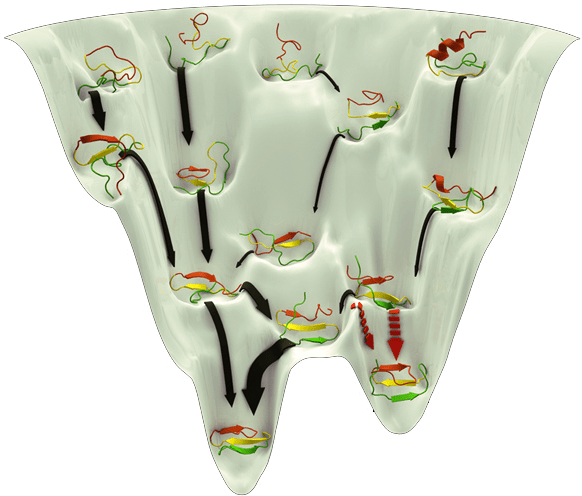
Levinthal’s Paradox: A protein can’t sample all conformations in a biologically reasonable time, yet it folds quickly
Example: A protein with 100 amino acids, each capable of adopting about 3 torsion angles, results in ~ possible conformations3100
3^{100}
What makes structure prediction hard: Complex energy landscape
Energy calculations are computationally intensive and depend on accurate force fields
Proteins fold to the lowest free-energy state, but this landscape is highly rugged
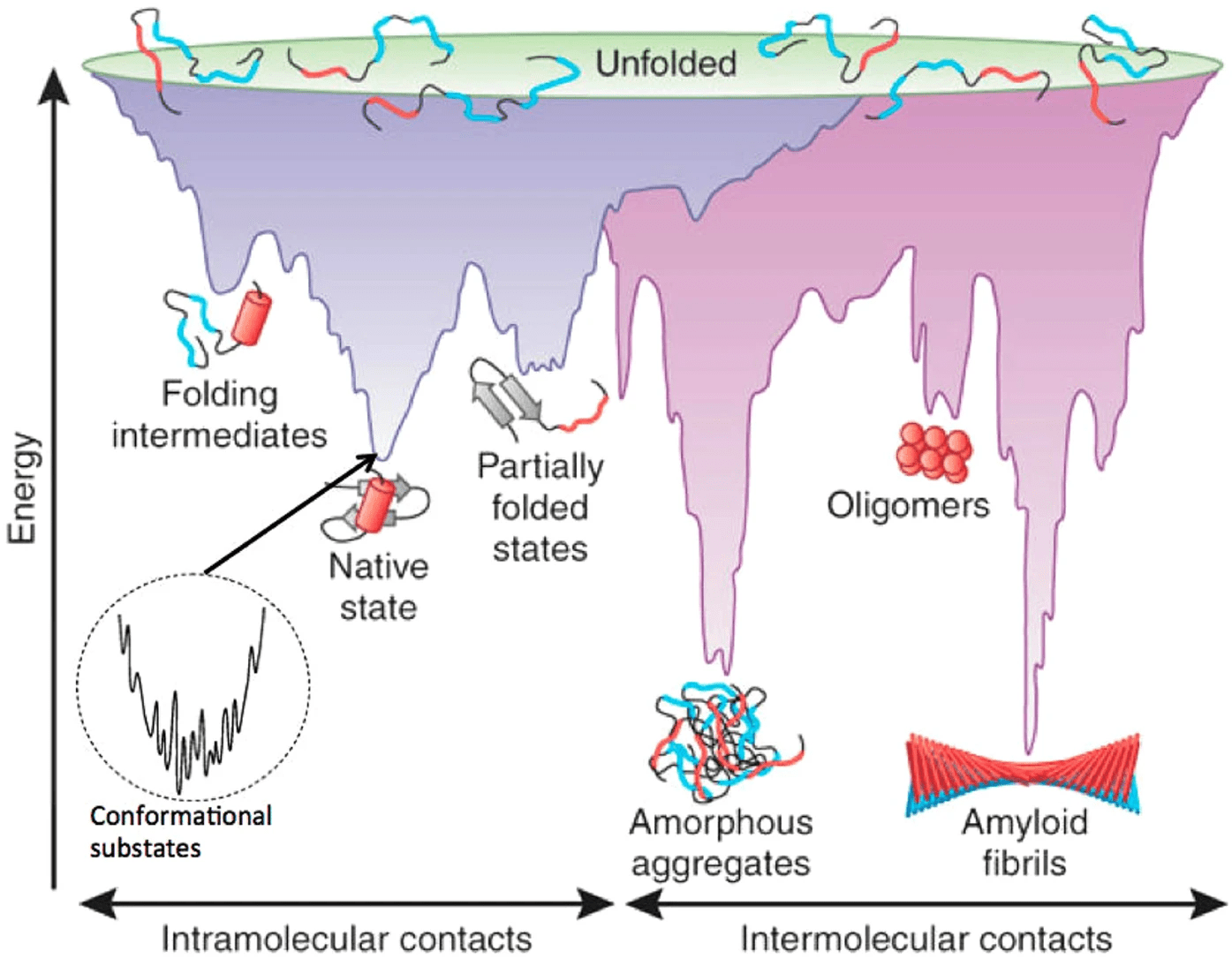
A potential energy surface (PES) is a represents the energy of a system as a function of the positions of its atoms
Understand how the system's energy changes upon reactions or movements
What makes structure prediction hard: Flexibility and dynamics
Proteins are not static; they adopt multiple conformations (flexibility) based on their environment or interactions with other molecules

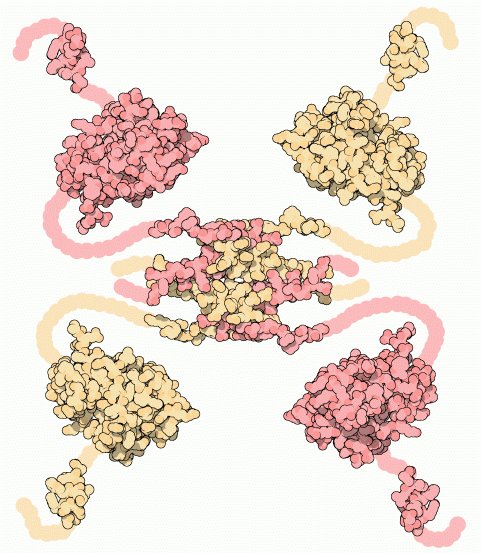
Some proteins or regions do not adopt a fixed 3D structure but remain disordered or flexible under physiological conditions
What makes structure prediction hard: Environmental effects
Proteins fold differently in different environments
Predictions need to capture interactions with solvent molecules, ions, and cofactors
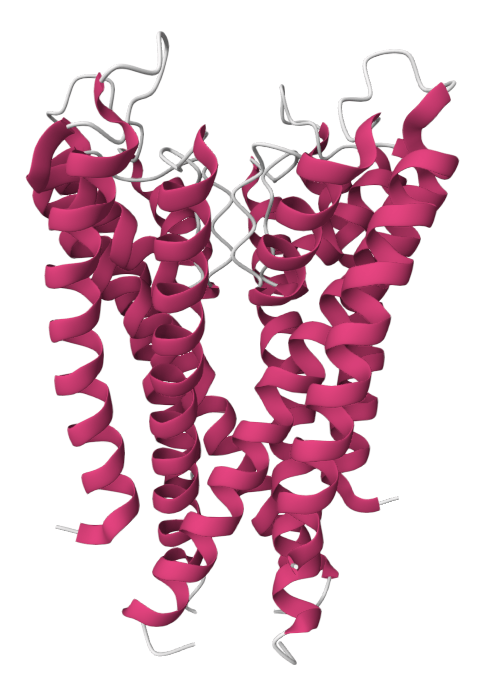
Example: Predicting transmembrane protein structures, where the lipid bilayer plays a key role in folding, is particularly complex.
AlphaFold 3
pH-gated K+ channel
What makes structure prediction hard: Post-translational modifications
PTMs such as phosphorylation, glycosylation, and methylation can alter protein folding and function
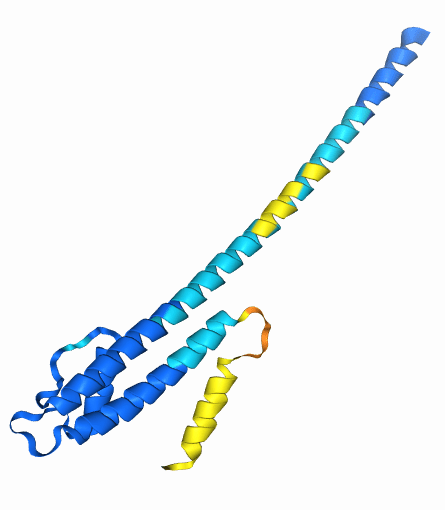
Example: eIF4E is a eukaryotic translation initiation factor involved in directing ribosomes to the cap structure of mRNAs
Ser209 is phosphorylated by MNK1
AlphaFold 3 accurately predicts these changes when they are already known
What makes structure prediction hard: Methods are data driven
Example: AlphaFold has made strides, but predicting de novo structures remains challenging, especially for proteins with no templates
Our predictions rely on similarity to known structures, but novel sequences or folds (for which no homologous structures exist) are difficult to predict accurately
After today, you should be able to
Explain homology modeling
Homology modeling predicts protein structures based on evolutionary relationships
Homology modeling is the most accurate when sequence identity to other proteins is high (>30%)
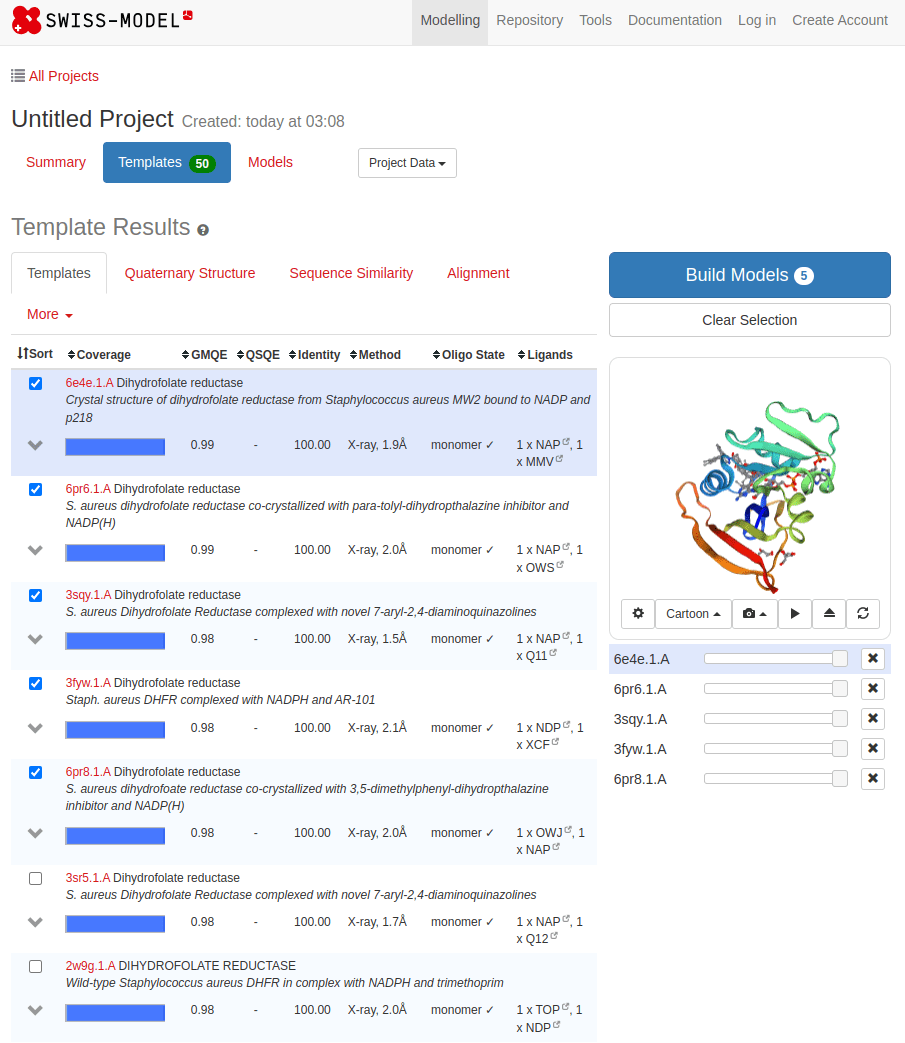
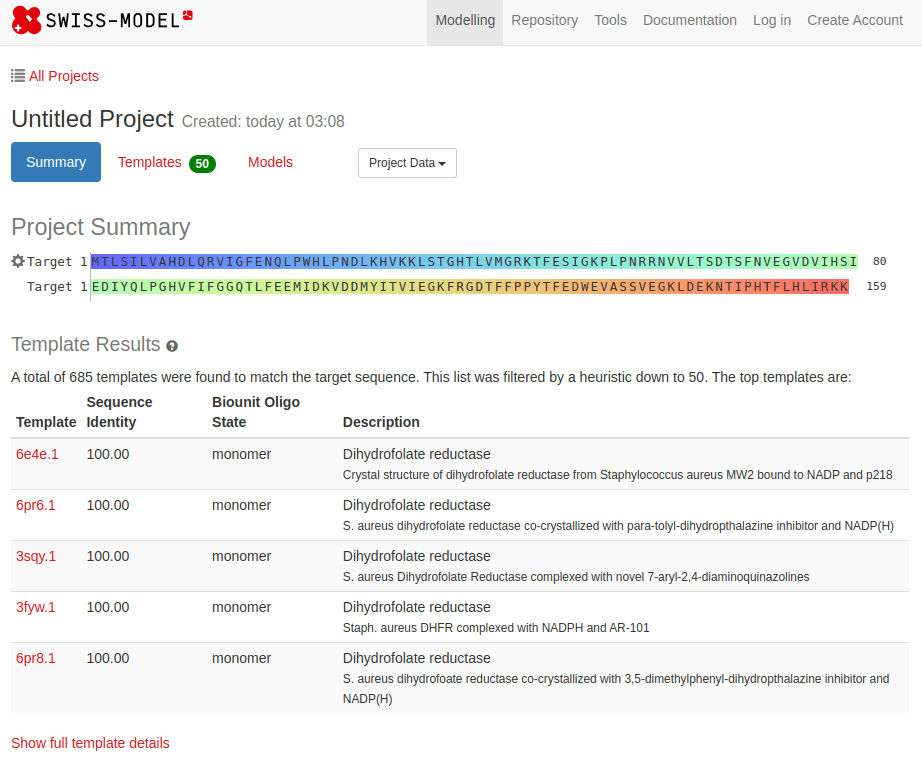
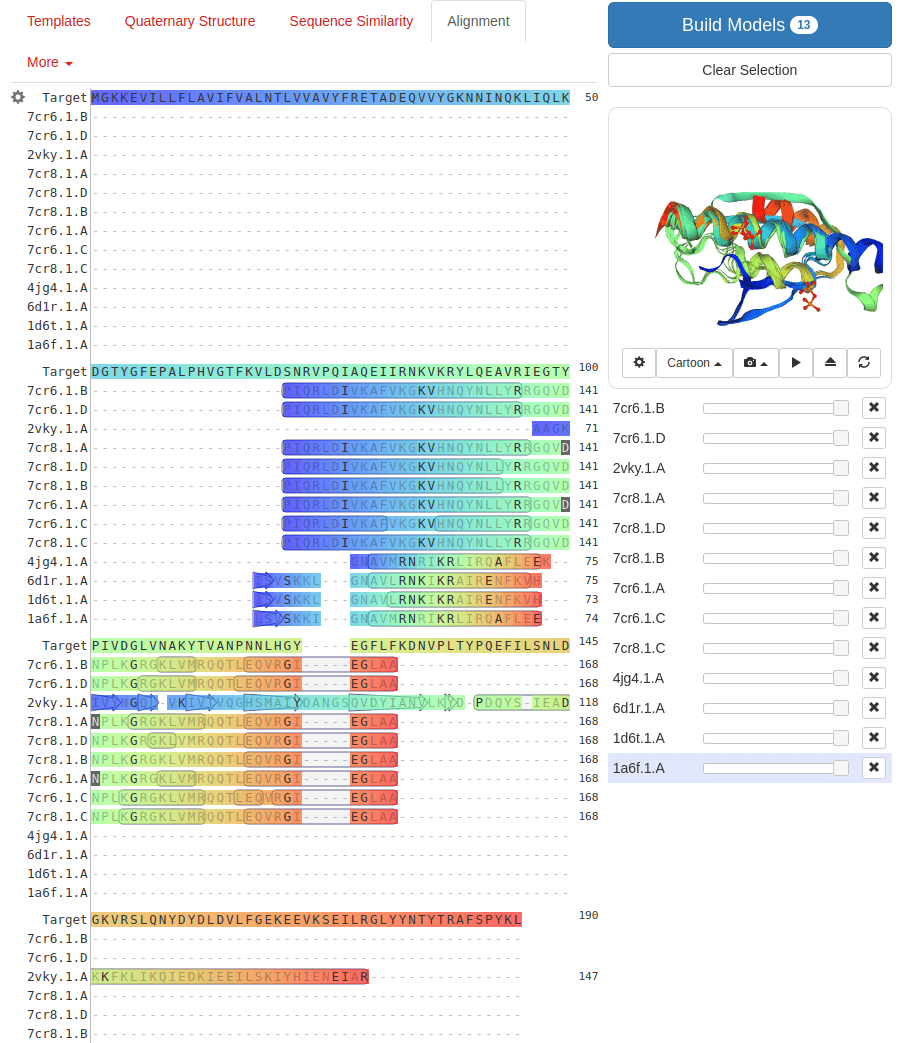
Common tools for homology modeling include MODELLER, SWISS-MODEL, and Phyre2
The main principle is that proteins with similar sequences tend to fold into similar structures
Hidden Markov Models (HMMs) Capture Evolutionary Patterns in Proteins
HMMs are statistical models representing sequences using probabilities for matches, insertions, and deletions
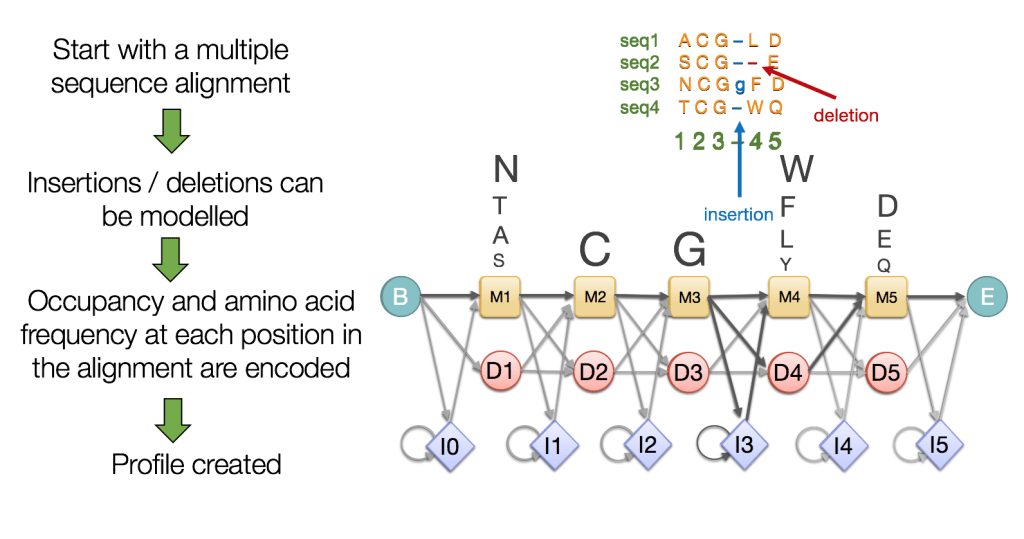
Essentially more robust alignments
A Markov model predicts outcomes based on transitional probabilities
Suppose I collect weather data in Pittsburgh for the past 30 days: Sunny, Cloudy, or Rain
I want to figure out how to predict tomorrow's weather based on today's
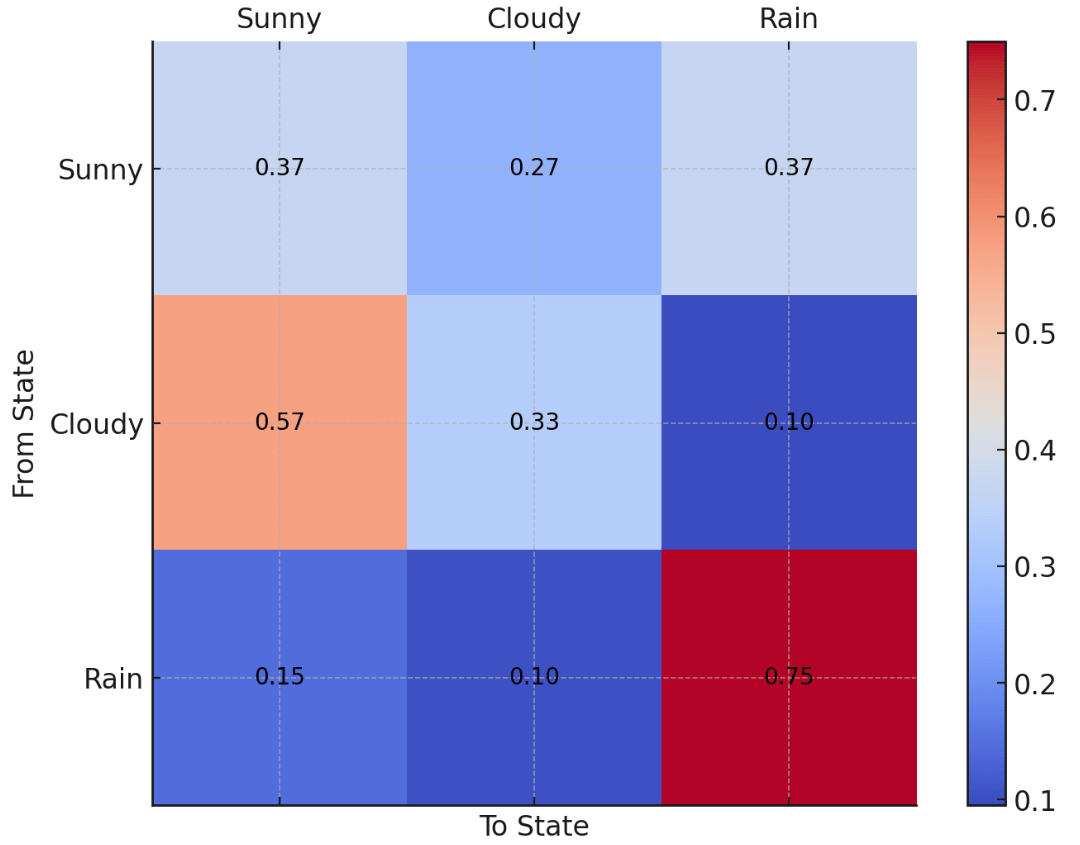
Today's weather
Tomorrow's weather
Transition probability
Example: If today is cloudy, there is a 57% chance it will be Sunny tomorrow
We can represent these states and probabilities as a (cursed?) graph
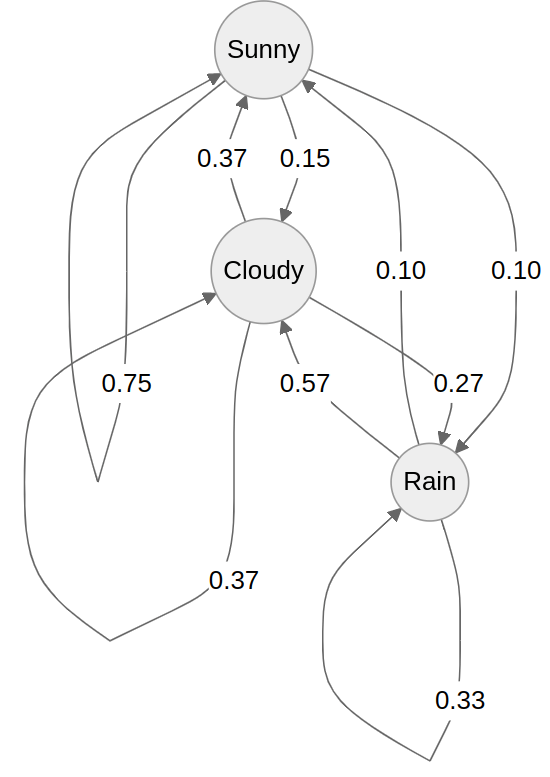
Each edge represents the probability of transitioning from one state to the next
Hidden Markov models also include additional information in "hidden states"
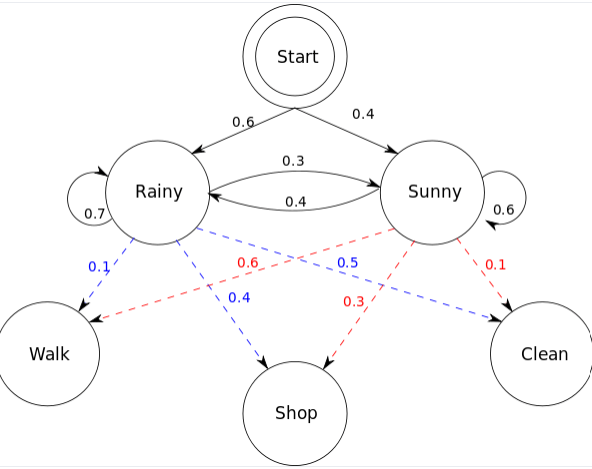
Suppose my friend lives in a remote location where it is either Rainy or Sunny
I cannot look up the weather but I have last year's weathers reports
- Walking
- Shopping
- Cleaning
We know how weather patterns transitions, but we don't have this information from our friend
Obervables
Hidden states
Note: If we had previous observable data, we could fit/learn transition probabilities of hidden states
My friend can only tell me
HMMs Model Protein Sequences as a Series of Probabilistic States
Hidden states represent the underlying biological events that are not directly observable
Observables are the actual amino acids (residues) in the protein sequence that we can observe
- Match states: conserved positions in the sequence
- Insertion states: positions where extra residues are added
- Deletion states: positions where residues are missing
HMMER Uses HMMs to Search Protein Databases for Homology
HMMER is a tool that uses HMMs to search databases for sequences that match a given profile HMM
It is used to find homologous sequences, identifying evolutionary relationships across protein families
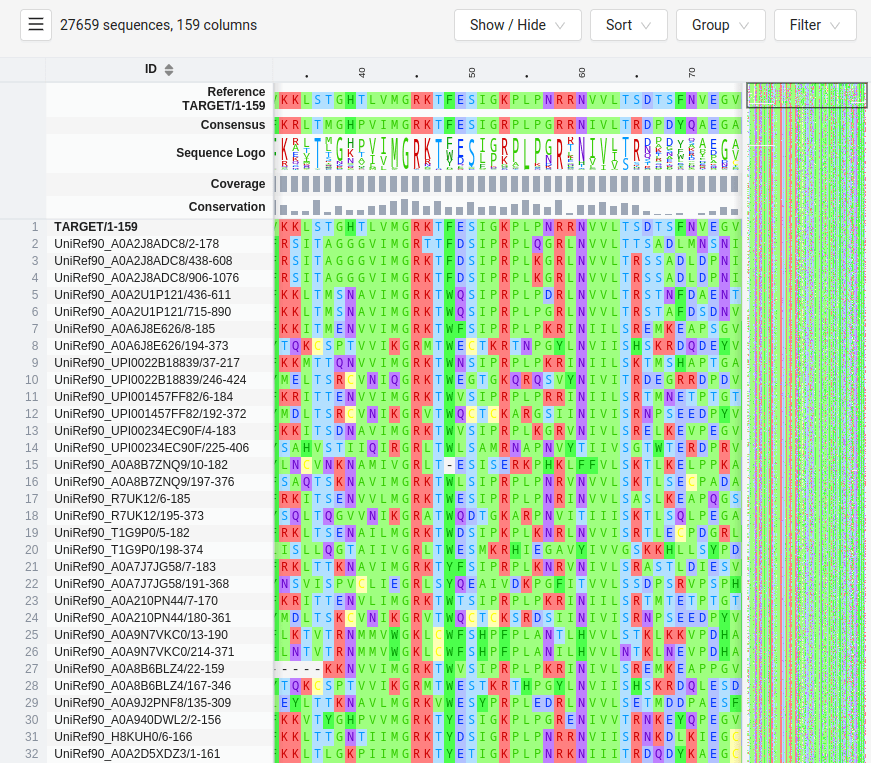
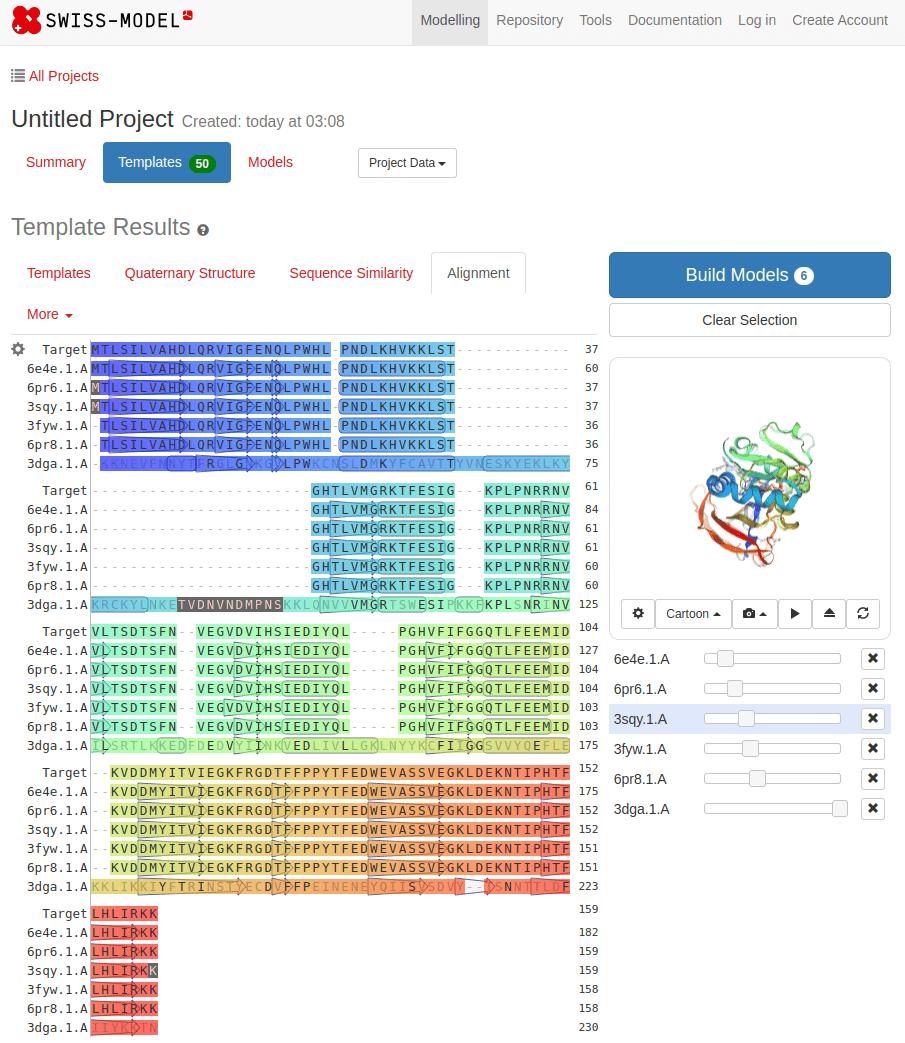
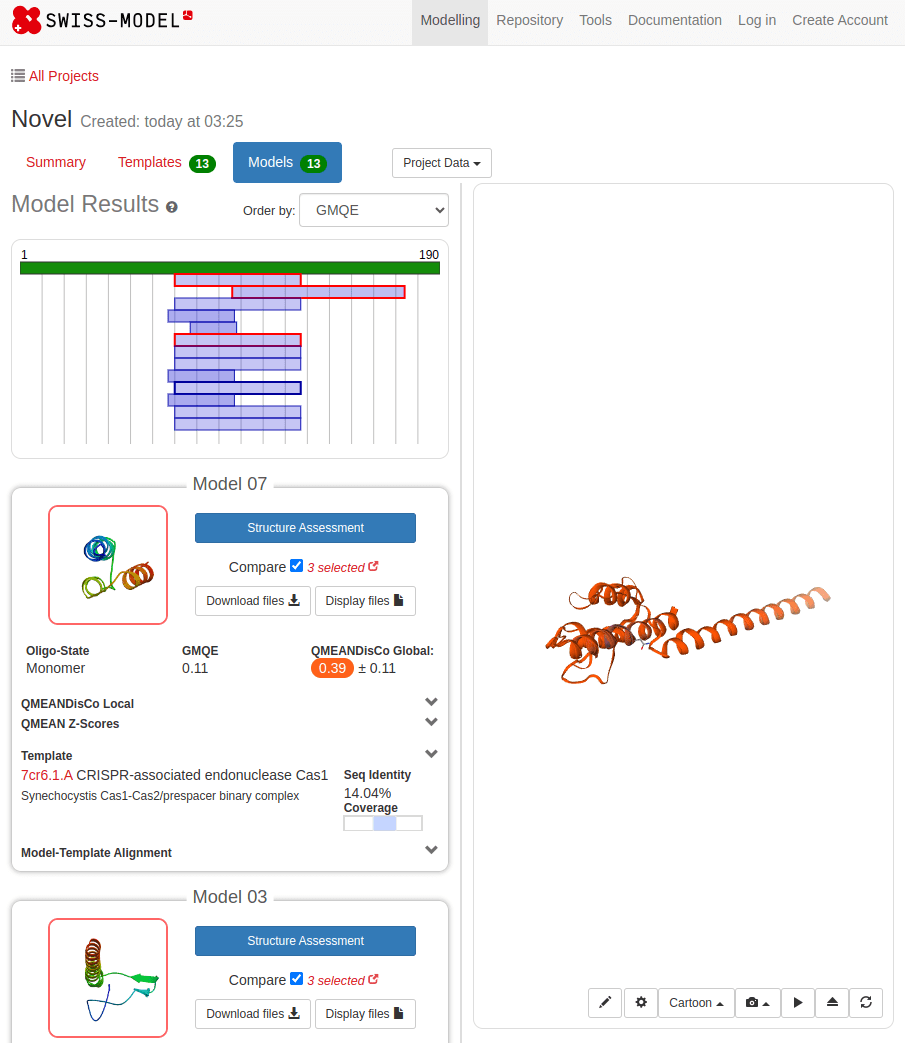
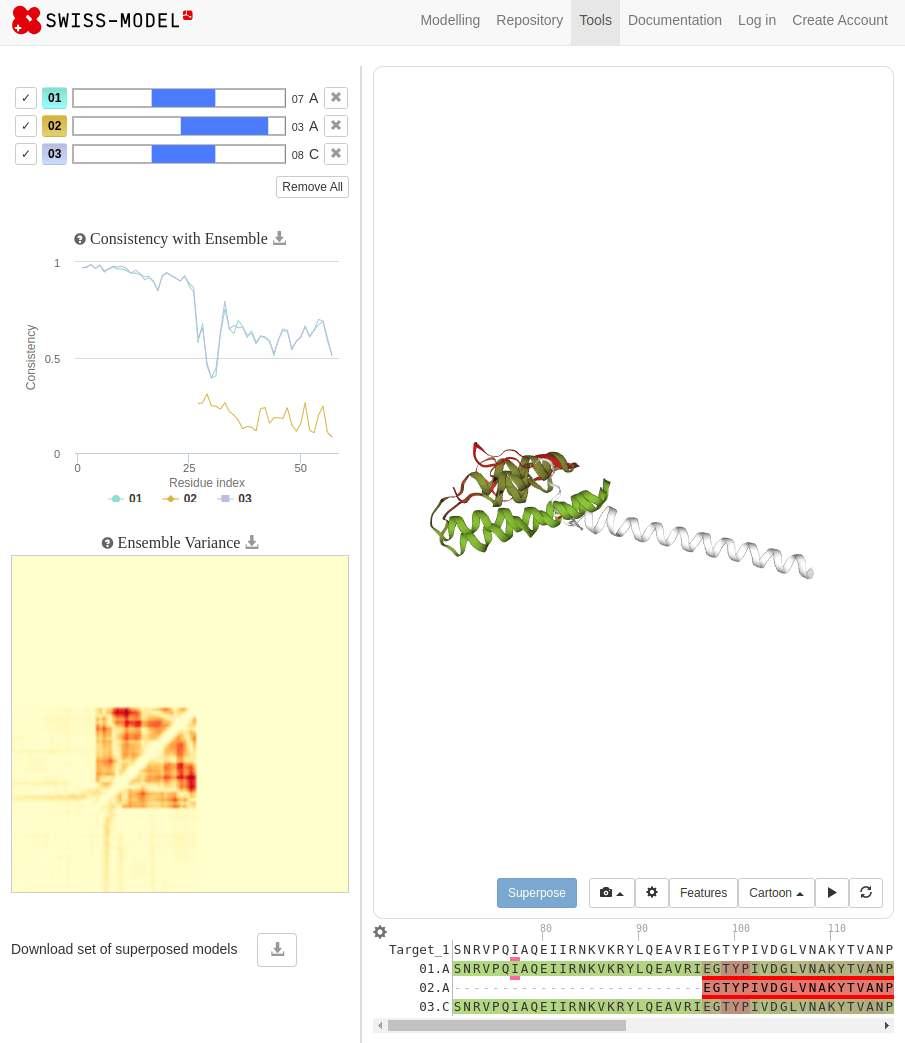
SWISS-MODEL
MTLSILVAHDLQRVIGFENQLPWHLPNDLKHVKKLSTGHTLVMGRKTFESIGKPLPNRRNVVLTSDTSFNVEGVDVIHSIEDIYQLPGHVFIFGGQTLFEEMIDKVDDMYITVIEGKFRGDTFFPPYTFEDWEVASSVEGKLDEKNTIPHTFLHLIRKKDHFR (UniProt)
SWISS-MODEL
SWISS-MODEL
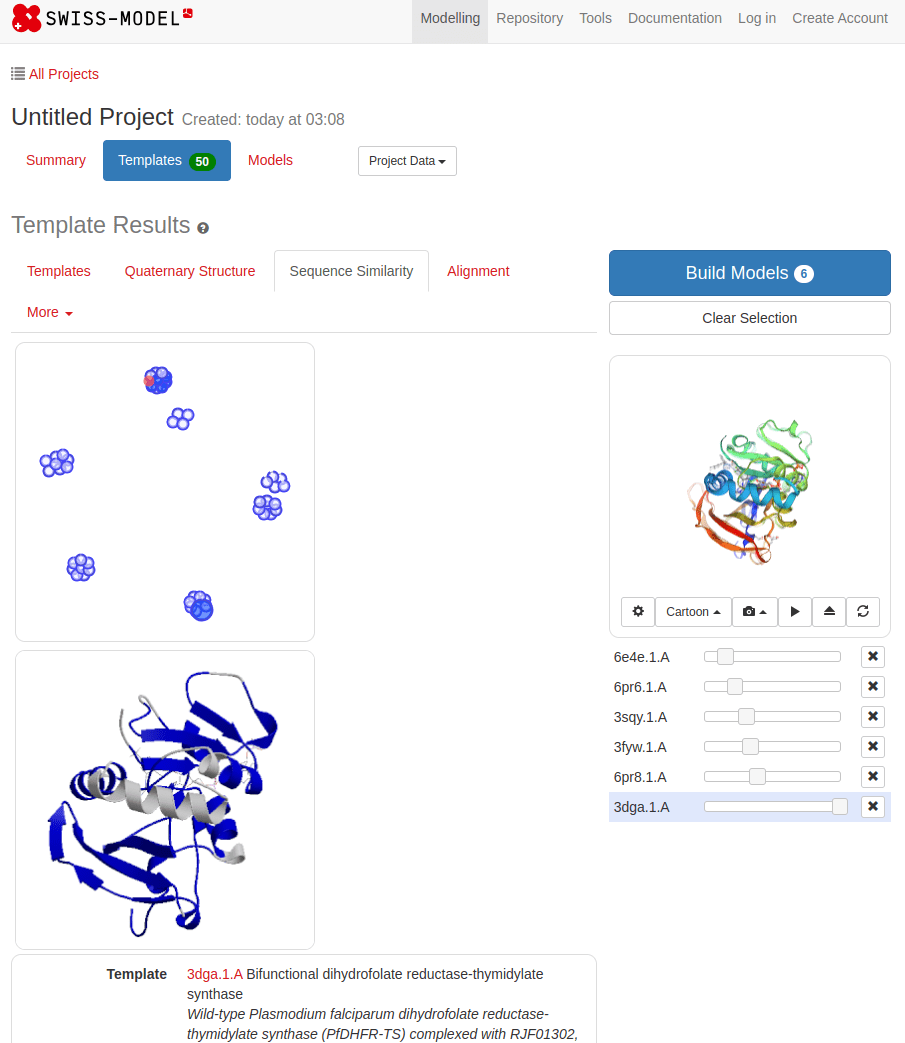
SWISS-MODEL
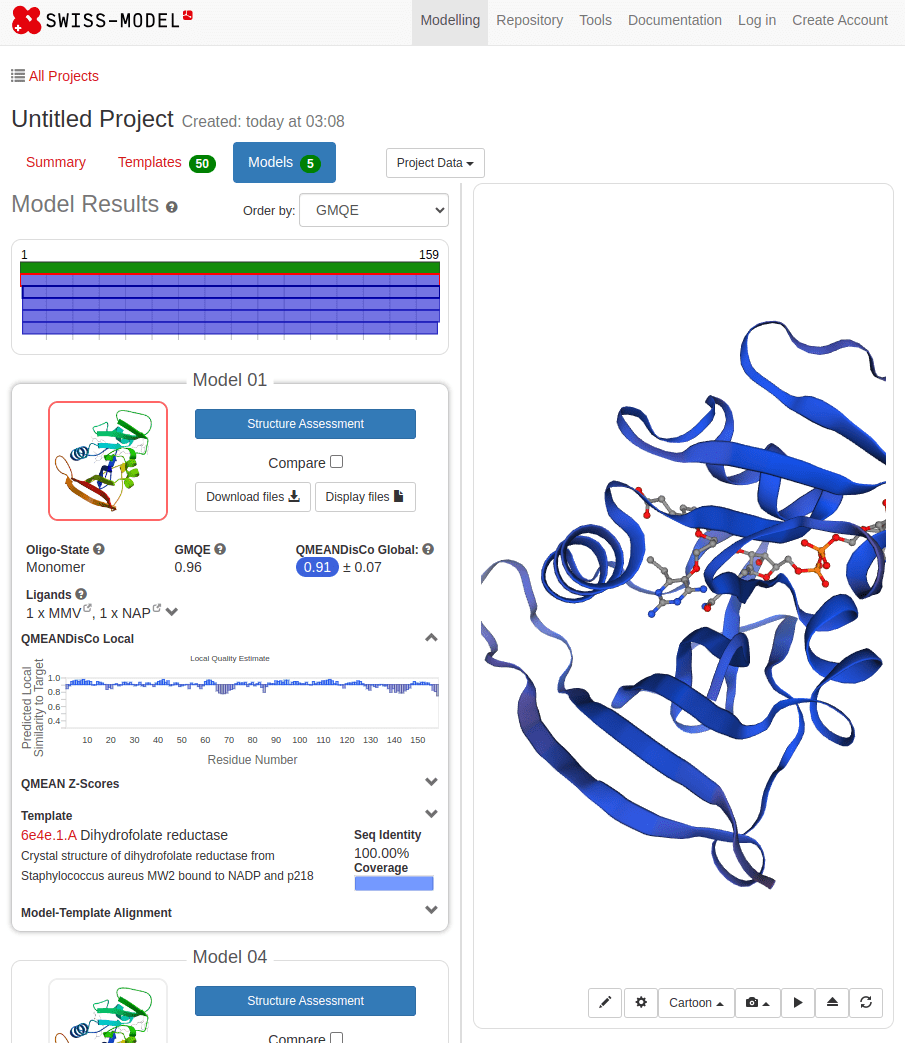
What happens with a novel protein?
MGKKEVILLFLAVIFVALNTLVVAVYFRETADEQVVYGKNNINQKLIQLKDGTYGFEPALPHVGTFKVLDSNRVPQIAQEIIRNKVKRYLQEAVRIEGTYPIVDGLVNAKYTVANPNNLHGYEGFLFKDNVPLTYPQEFILSNLDGKVRSLQNYDYDLDVLFGEKEEVKSEILRGLYYNTYTRAFSPYKLNovel protein (ChatGPT)
Novel proteins are too challenging
After today, you should be able to
Know when to use threading instead of homology modeling
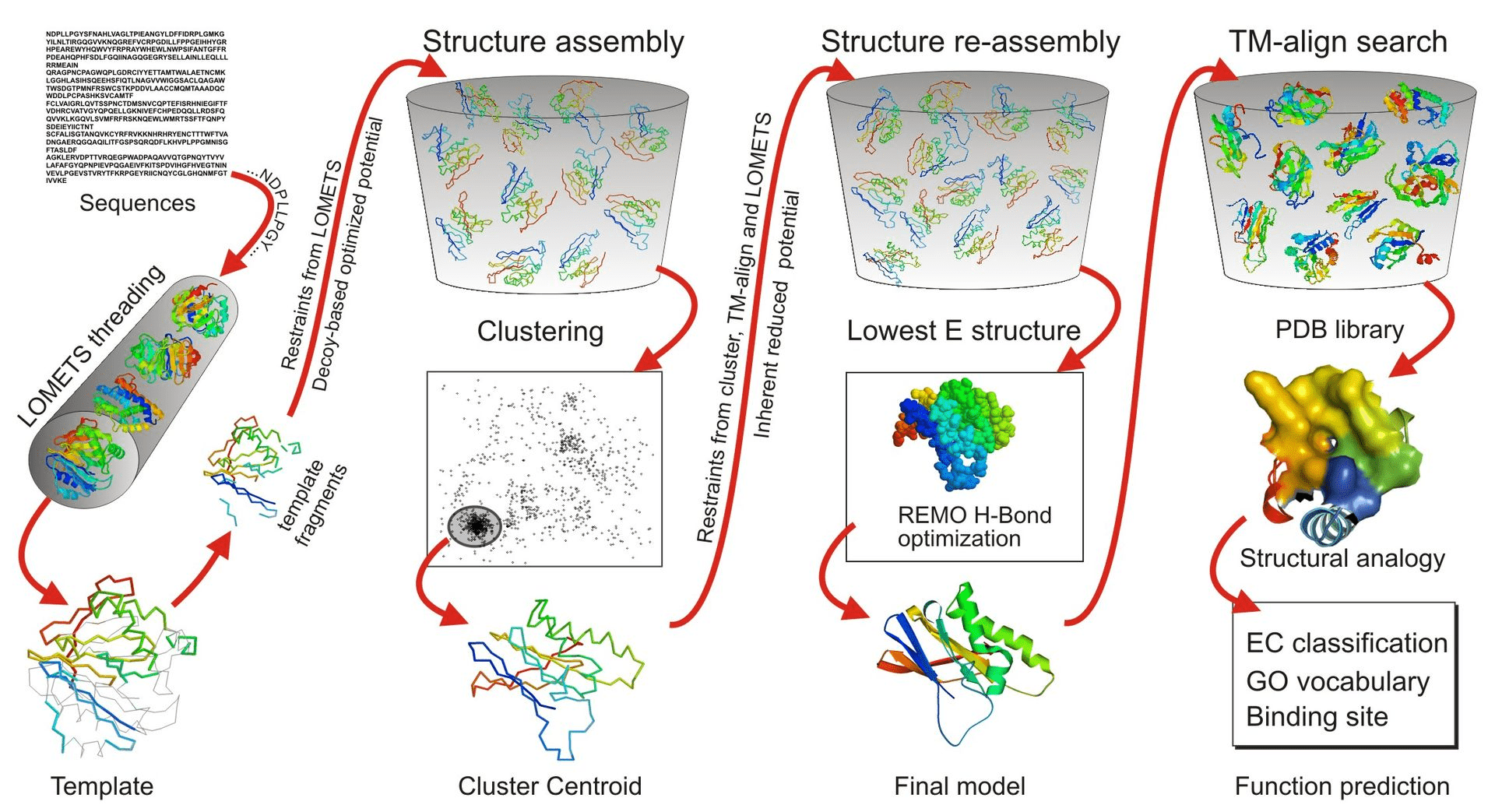
Why Use Threading?
In cases where sequence similarity to known structures is low (< 30%), homology modeling becomes unreliable
Phyre2, RaptorX, MUSTER, and I-TASSER are commonly used for threading and takes much longer than homology modeling
Threading matches sequences to known structural folds based on structural rather than sequence similarity
Identifying the Right Fold
After today, you should be able to
Interpret a contact map for protein structures
Contact Maps Visualize Residue Interactions in Proteins
A contact map is a 2D representation of which residues are in close proximity
Each point on the map corresponds to two residues that are close in 3D space
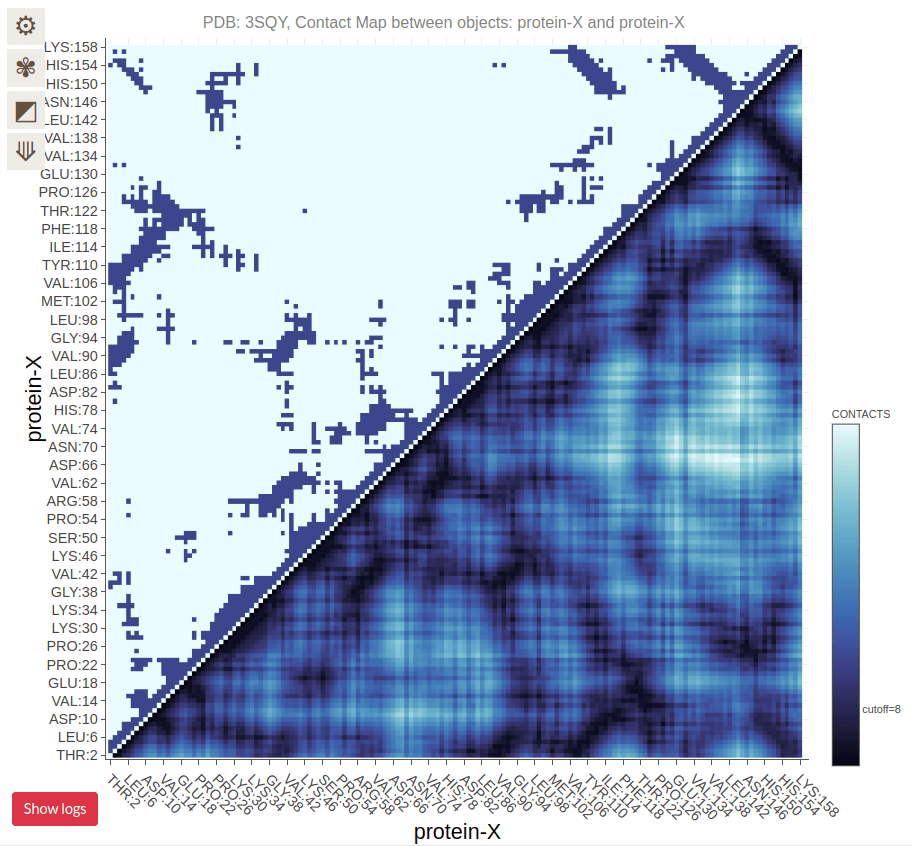
Contact Maps Represent Spatial Proximity, Not Sequence Order
Contacts are determined by spatial proximity, typically within a certain distance threshold
Residues far apart in the sequence can still be close in the 3D structure, reflected in the contact map
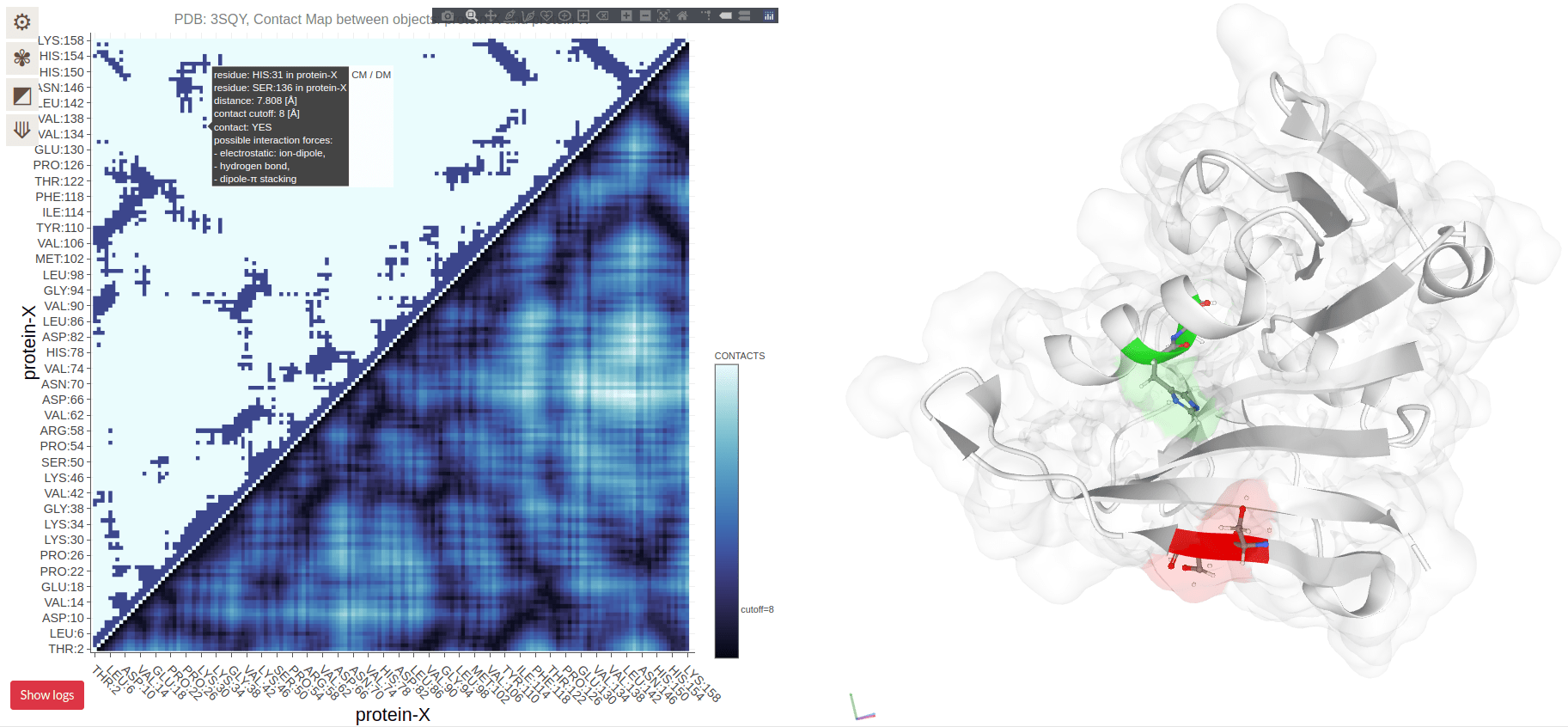
Residues on the diagonal are adjacent in sequence (and spatially)

After today, you should be able to
Comprehend how coevolution provides structural insights
The Rise of Machine Learning in Structural Biology
Traditional methods like homology modeling and threading rely on templates and known structures
AlphaFold (DeepMind) and RosettaFold (Baker Lab) lead the charge in this area
ML predicts 3D structures only from sequence data
What is AlphaFold?
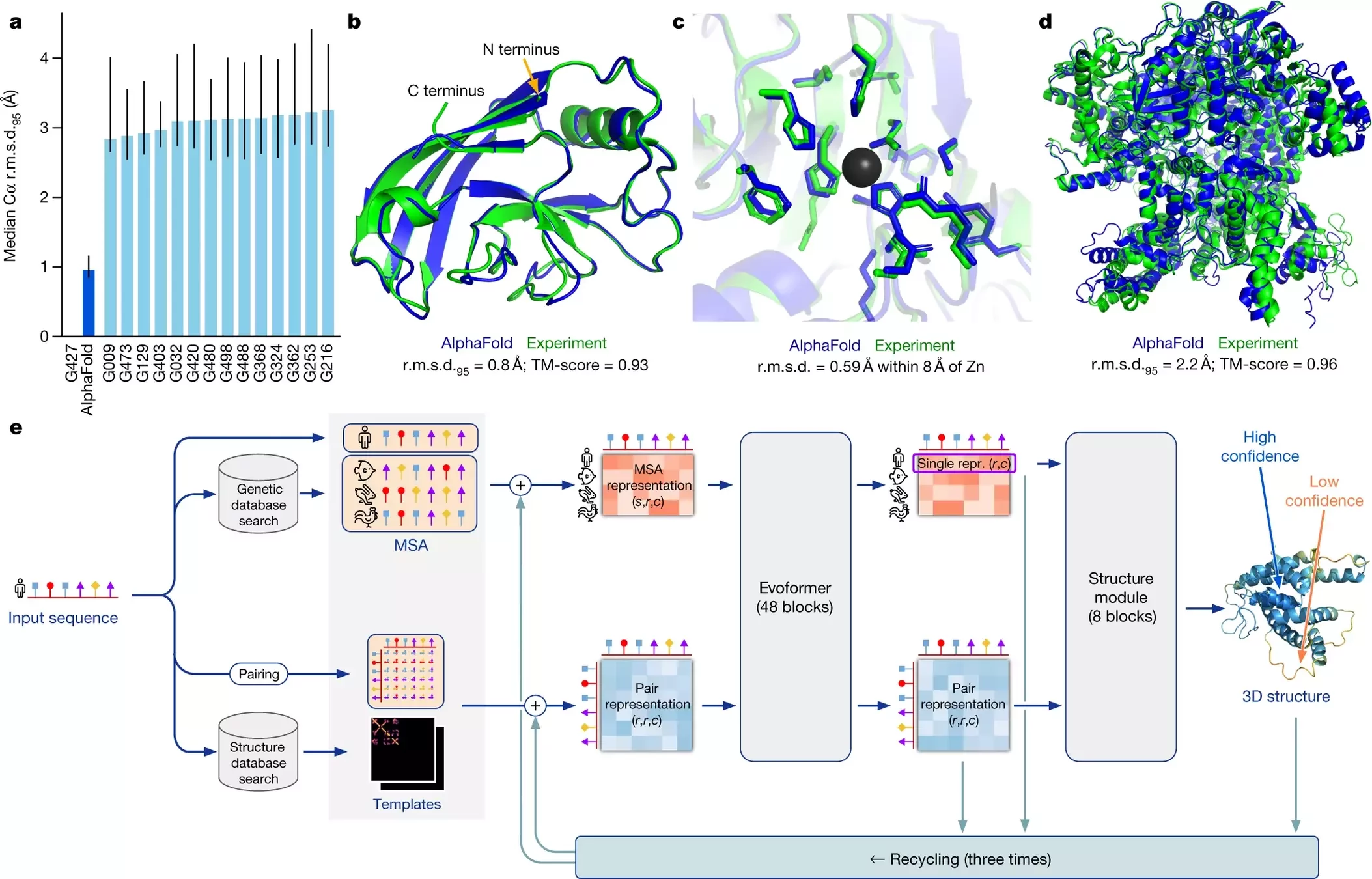
Developed by DeepMind, AlphaFold predicts protein structures with atomic accuracy by using deep learning models trained on large structural datasets
Breakthroughs
- AlphaFold 2 achieved near-experimental level accuracy in the 2020 CASP14 competition (Critical Assessment of protein Structure Prediction)
- AlphaFold 3 (2024) predicts proteins, DNA, RNA, ligands, and post-translational modifications
Coevolving residues mutate in a correlated manner
Mutations in one residue often result in compensatory mutations in its interacting partner
This is observed across species through analysis of homologous protein sequences
Correlated mutations indicate functionally significant residue pairs
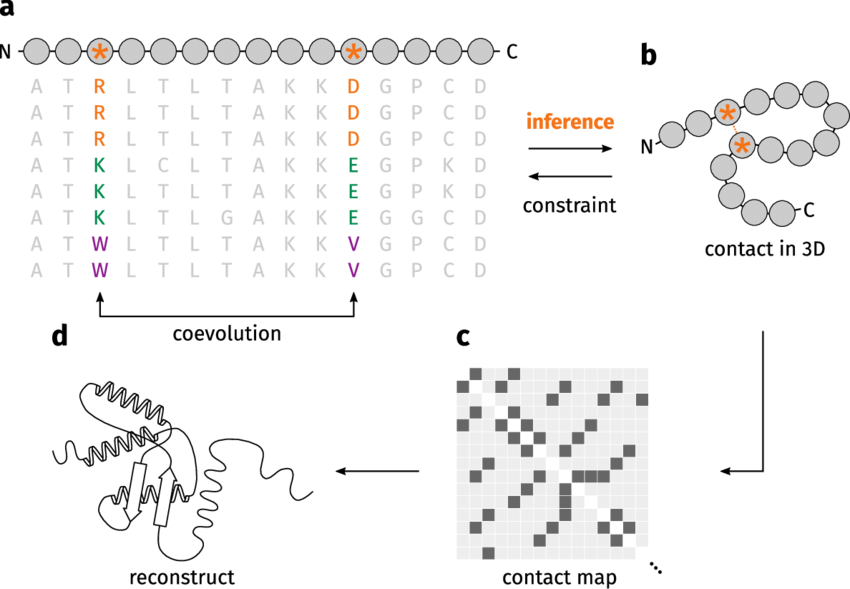
Arg (positive)
Asp (negative)
Lys (positive)
Glu (negative)
Trp (hydrophobic)
Val (hydrophobic)
Evolution
Evolutionary Analysis Reveals Structural Insights
Coevolution analysis helps predict which residues are close in the 3D structure
Residues showing correlated mutations are likely to be spatially close in the folded protein
This is particularly useful when no experimental structure is available
Multiple Sequence Alignments Enable Coevolution Detection
Coevolution is detected using large MSAs from homologous proteins
The more diverse the sequences in the MSA, the better the resolution of coevolving residues
Evolutionary information from MSAs guides predictions for residue-residue contacts
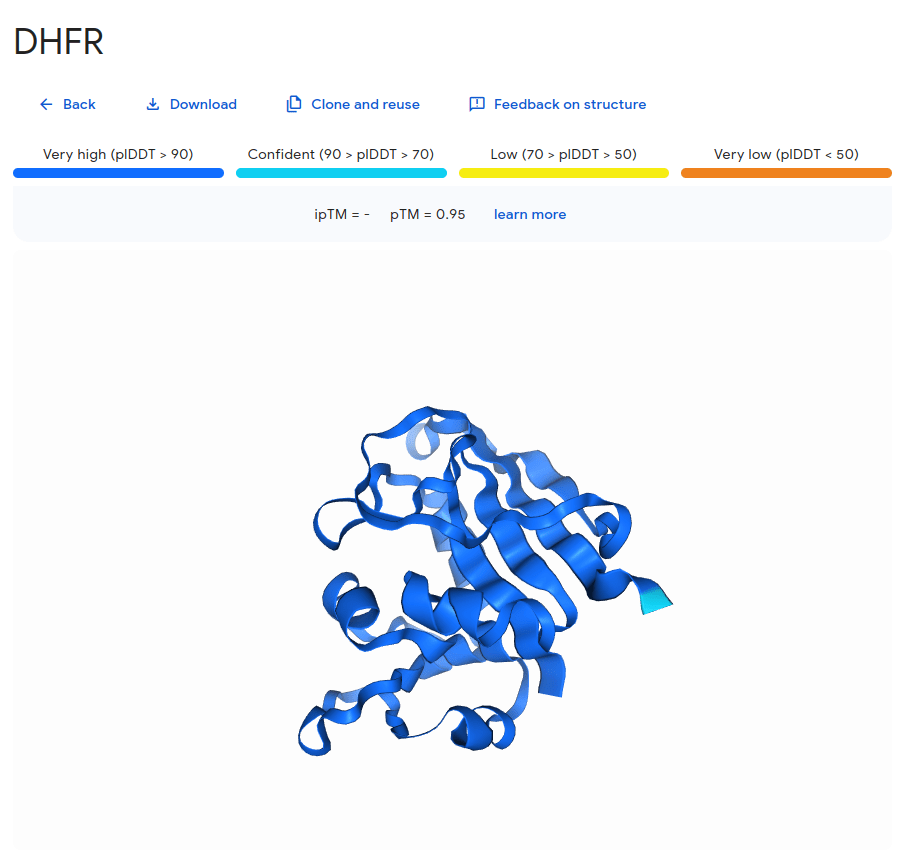
Coevolution example: DHFR
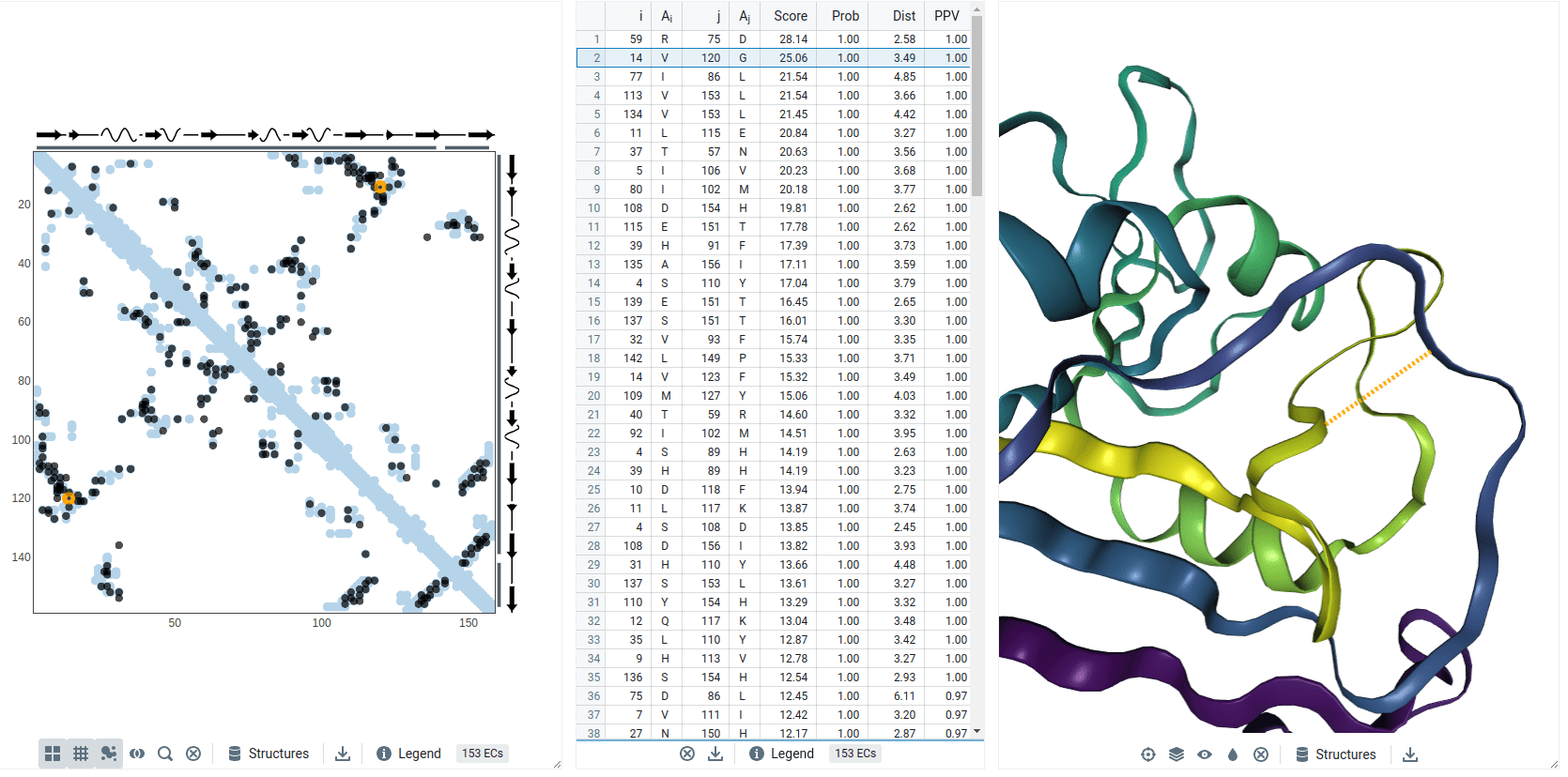
Residues with a high Score (i.e., coevolve) are near each other in the protein's structure (i.e., small distance)
Val14 and Gly120 coevolved
Models predict these residues are spatially close
Coevolutionary signals can be noisy
Not all correlated mutations are due to direct physical interactions; some may be indirect
Noise in the data can come from random mutations or insufficient evolutionary diversity.
Large and diverse sequence data sets are needed for reliable coevolution predictions.
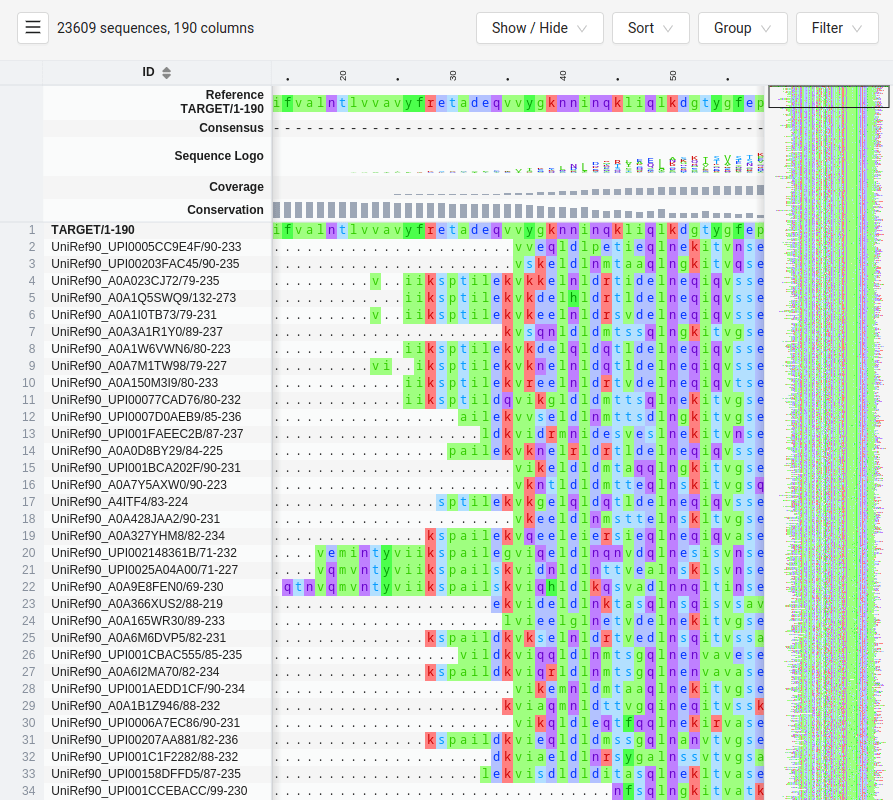
Machine learning leverages coevolution for high-accuracy predictions
AlphaFold and RosettaFold utilize coevolutionary data from MSAs to predict residue interactions
These models incorporate evolutionary information along with structural features, leading to highly accurate predictions
After today, you should be able to
Explain why ML models are dominate protein structure prediction
AlphaFold pipeline, simplified
Given the following data
MTLSILVAHDLQRVIGFENQLPWHLPNDLKHVKKLSTGHTLVMGRKTFESIGKPLPNRRNVVLTSDTSFNVEGVDVIHSIEDIYQLPGHVFIFGGQTLFEEMIDKVDDMYITVIEGKFRGDTFFPPYTFEDWEVASSVEGKLDEKNTIPHTFLHLIRKKInput sequence
Multiple Sequence Alignment
Predict
Atomistic structure
ML models
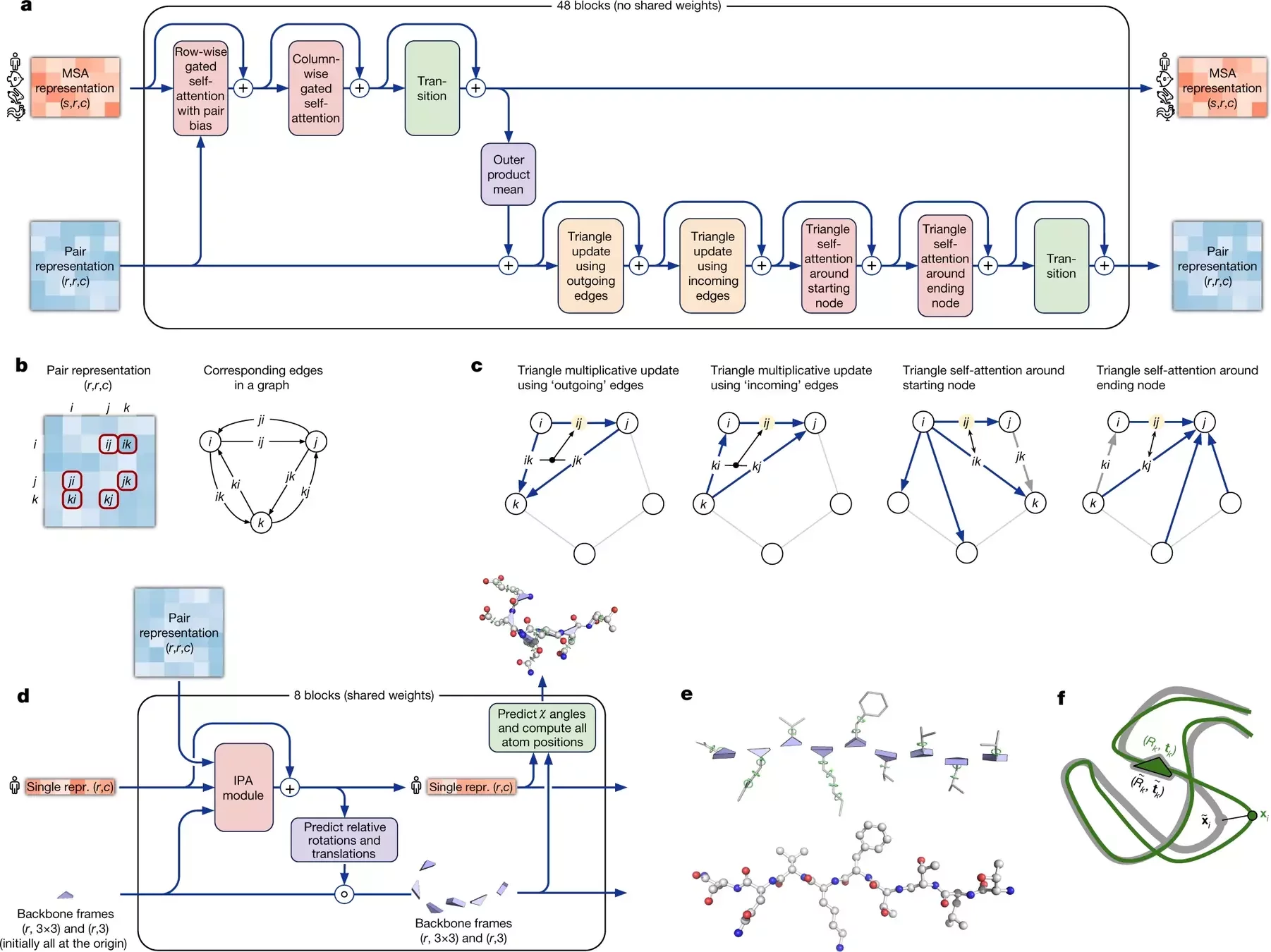
AlphaFold 2 pipeline: Evoformer
Using MSAs and contact maps, DeepMind trained a model to predict protein structures
Contact maps are converted into dihedral angles
What is new in AlphaFold 3?
Biggest change is the use of a diffusion model
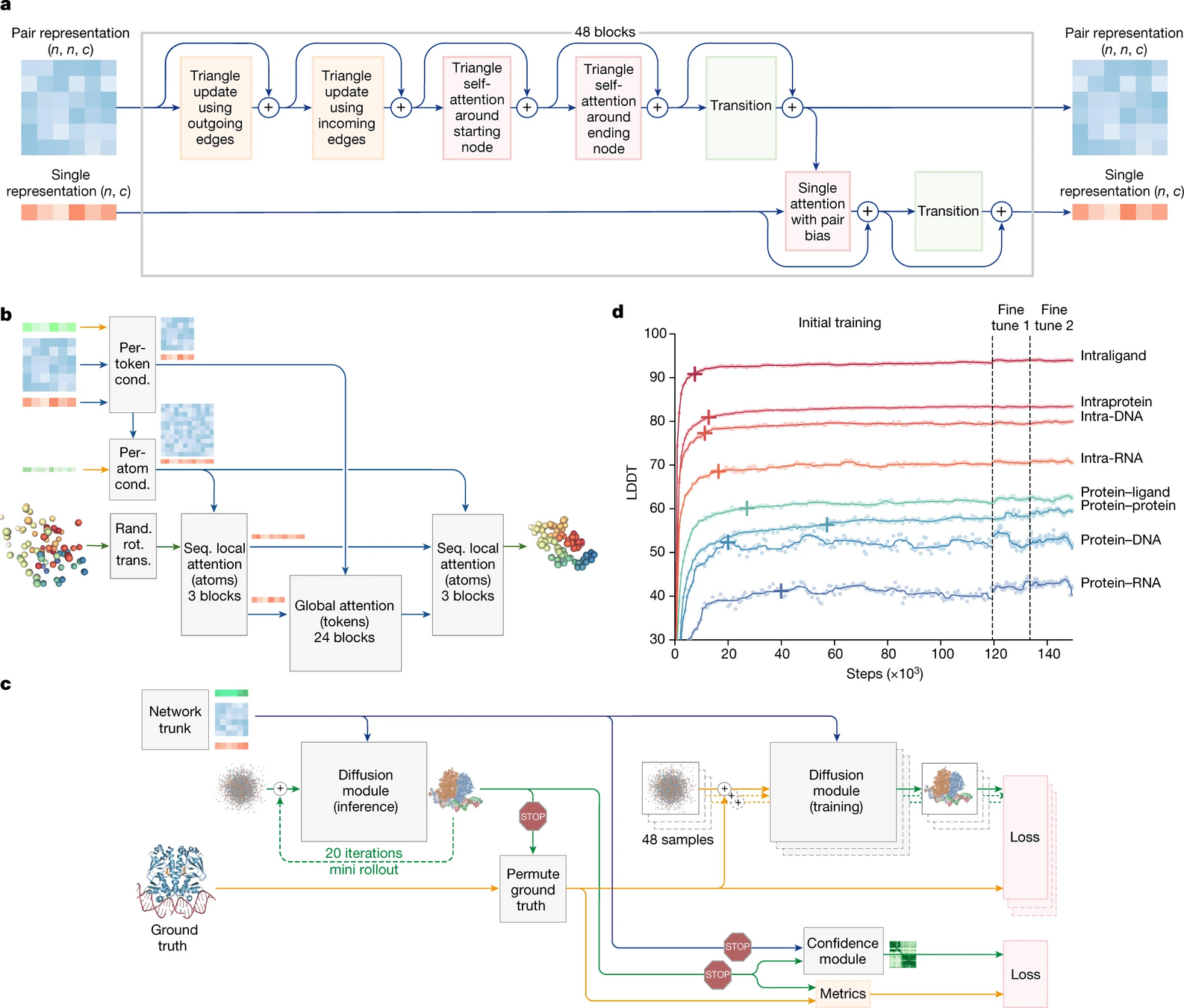
Diffusion models essentially learn to unscramble atoms into a structure
AlphaFold 3 is supercharged for any biomolecule
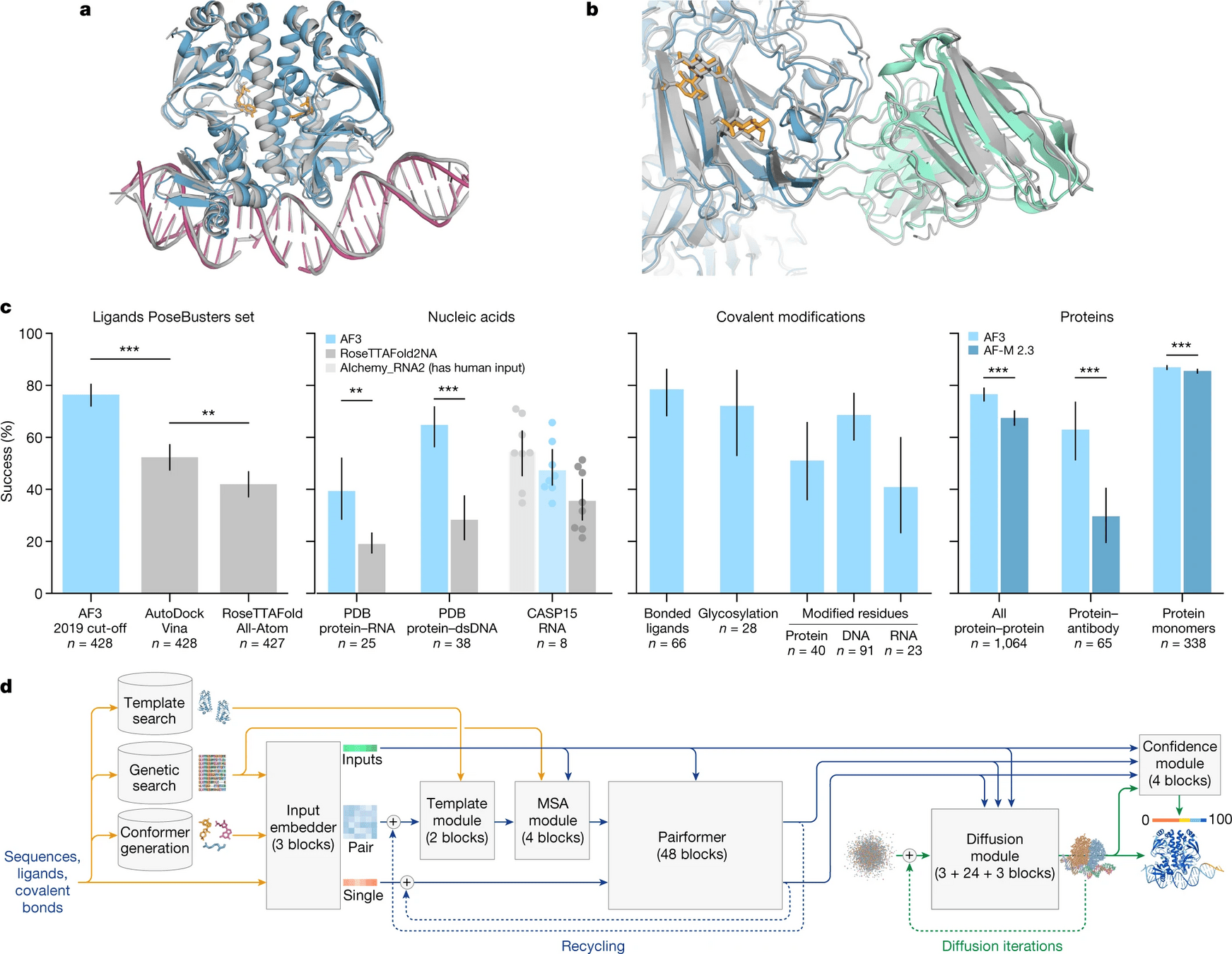
Proteins, DNA, RNA, ligands, PTMs, protein-proteins, etc.
AlphaFold 3
MTLSILVAHDLQRVIGFENQLPWHLPNDLKHVKKLSTGHTLVMGRKTFESIGKPLPNRRNVVLTSDTSFNVEGVDVIHSIEDIYQLPGHVFIFGGQTLFEEMIDKVDDMYITVIEGKFRGDTFFPPYTFEDWEVASSVEGKLDEKNTIPHTFLHLIRKKDHFR (UniProt)
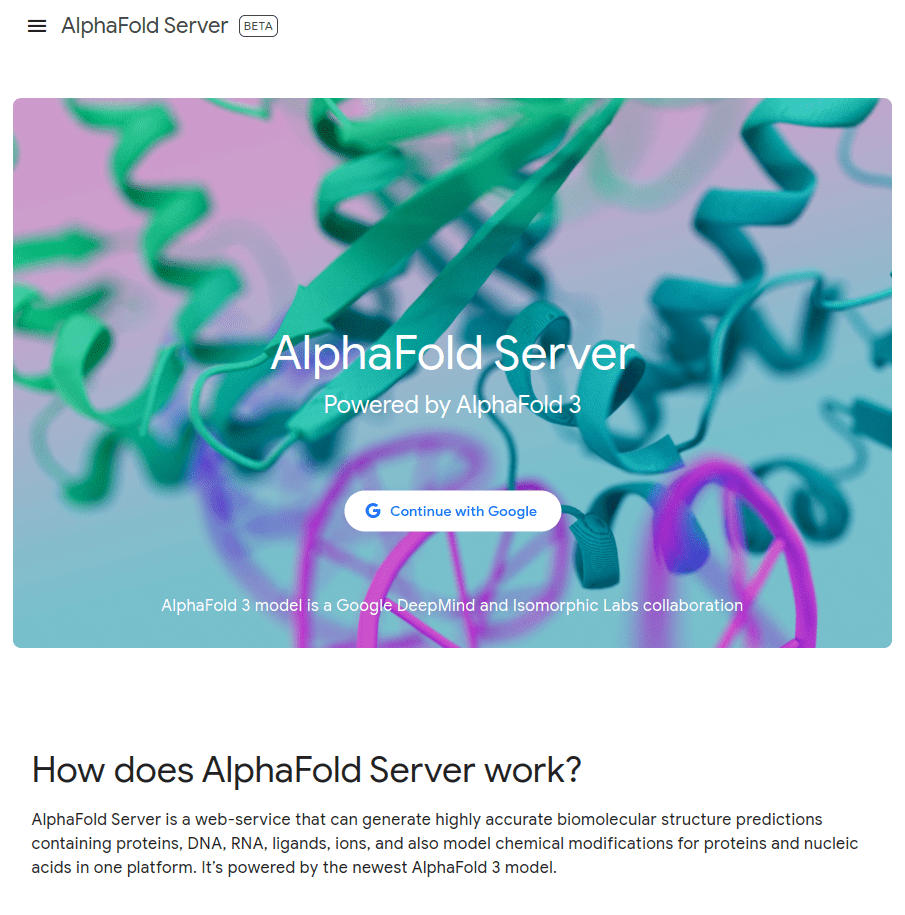
MGKKEVILLFLAVIFVALNTLVVAVYFRETADEQVVYGKNNINQKLIQLKDGTYGFEPALPHVGTFKVLDSNRVPQIAQEIIRNKVKRYLQEAVRIEGTYPIVDGLVNAKYTVANPNNLHGYEGFLFKDNVPLTYPQEFILSNLDGKVRSLQNYDYDLDVLFGEKEEVKSEILRGLYYNTYTRAFSPYKLNovel protein (ChatGPT)
AlphaFold 3 is a breakthrough, not the final solution
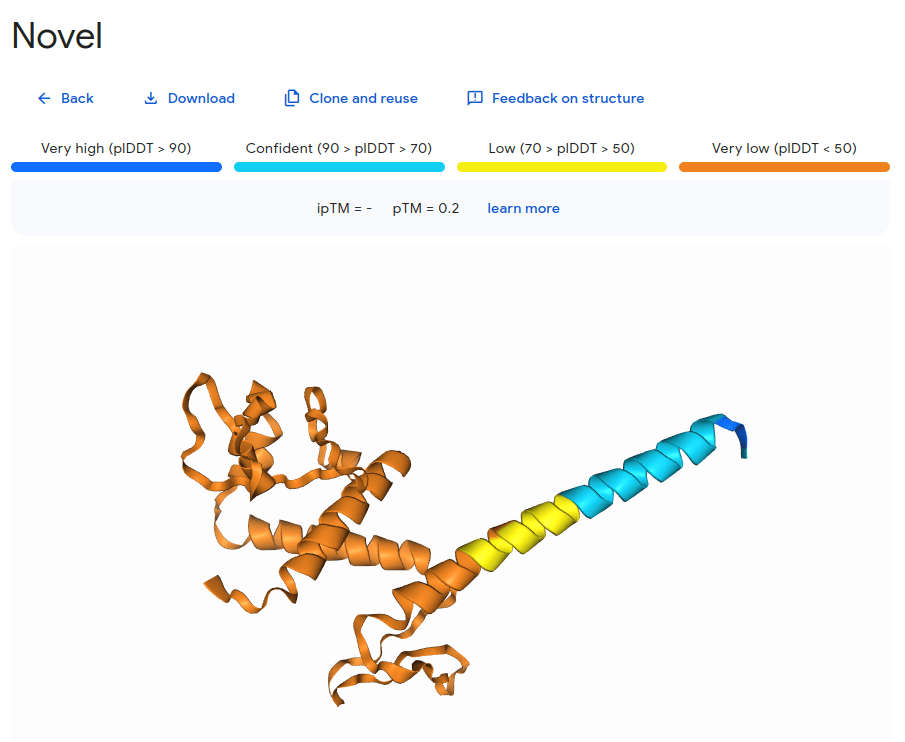
Caveat: Proteins are dynamic
What about intrinsically disordered proteins?
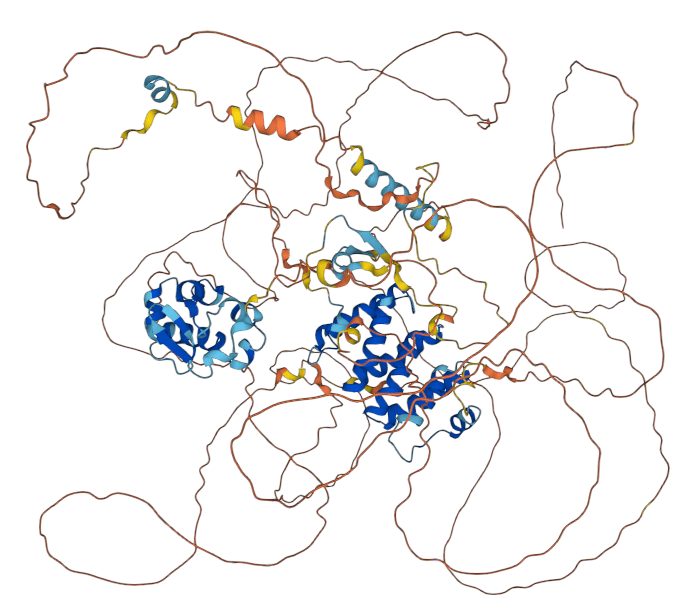
At least 40% of proteins have disordered regions
AlphaFold (and all other methods) struggle with disordered regions
Before the next class, you should
- Work on A05
- Review material
Lecture 12:
Protein structure prediction
Today
Thursday
Lecture 13:
Molecular simulation princples
BIOSC 1540: L12 (Protein structure prediction)
By aalexmmaldonado



