Computational Biology
(BIOSC 1540)
Oct 22, 2024
Lecture 14:
Molecular system representations
Announcements
- A05 is due Thursday by 11:59 pm
- A06 will be posted on Friday

After today, you should be able to
Explain why DHFR is a promising drug target.
THF production is crucial for cellular growth
THF is needed for
- Producing red blood cells,
- Synthesizing purines,
- Interconverting amino acids,
- Methylating tRNA,
- Generating and using formate.
Disrupting THF production has a cascading effect on essential cellular processes, primarily affecting DNA and RNA synthesis and amino acid metabolism
This is a useful process for drug design
DHFR is responsible for synthesizing THF
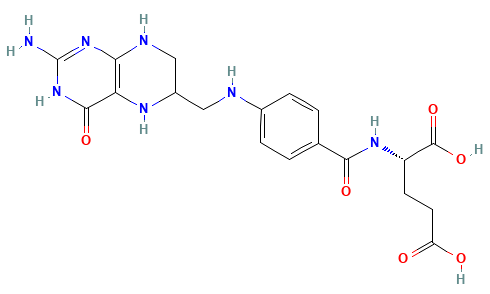
Dihydrofolate reductase (DHFR) is a crucial enzyme that produces THF from dihydrofolate (DHF)
DHF + NADPH
THF + NADP(+)
DHF
NADPH
(We will use this protein for our project)
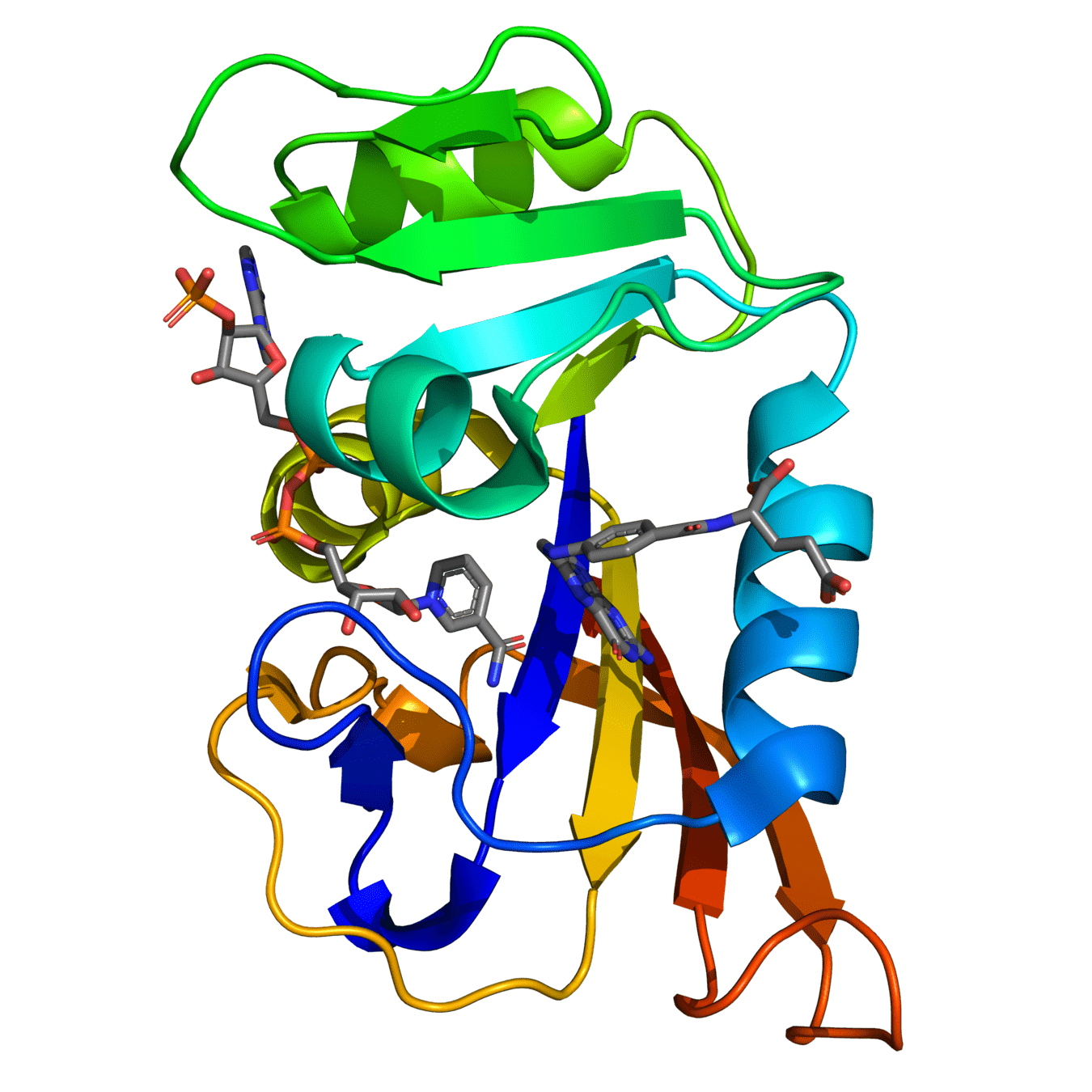
DHFR has been extensively studied as an antibiotic (e.g., trimethoprim) and cancer (e.g., methotrexate) target
DHFR conservation complicates drug design
Patient could have deleterious side effects
What would happen if a patient with a bacterial infection is prescribed a drug loosely targeting DHFR
Both proteins have high structural similarity, even around the active site
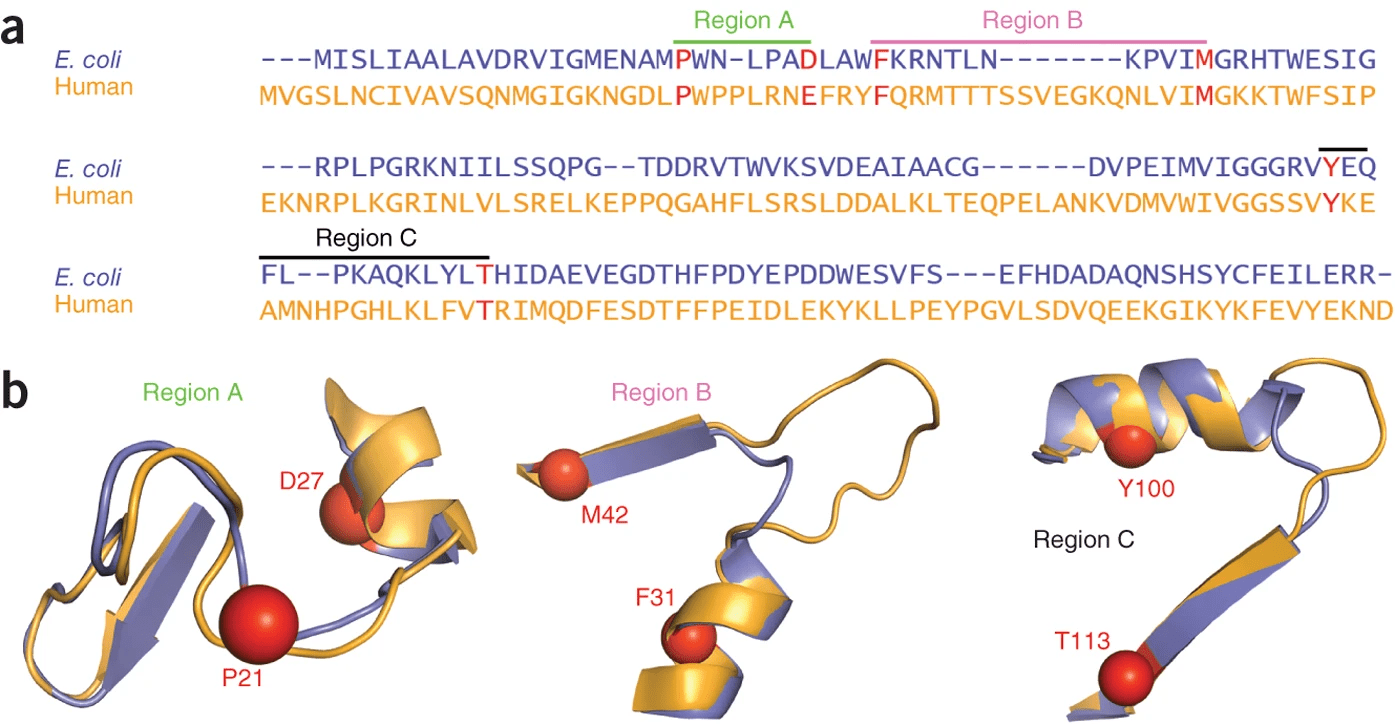
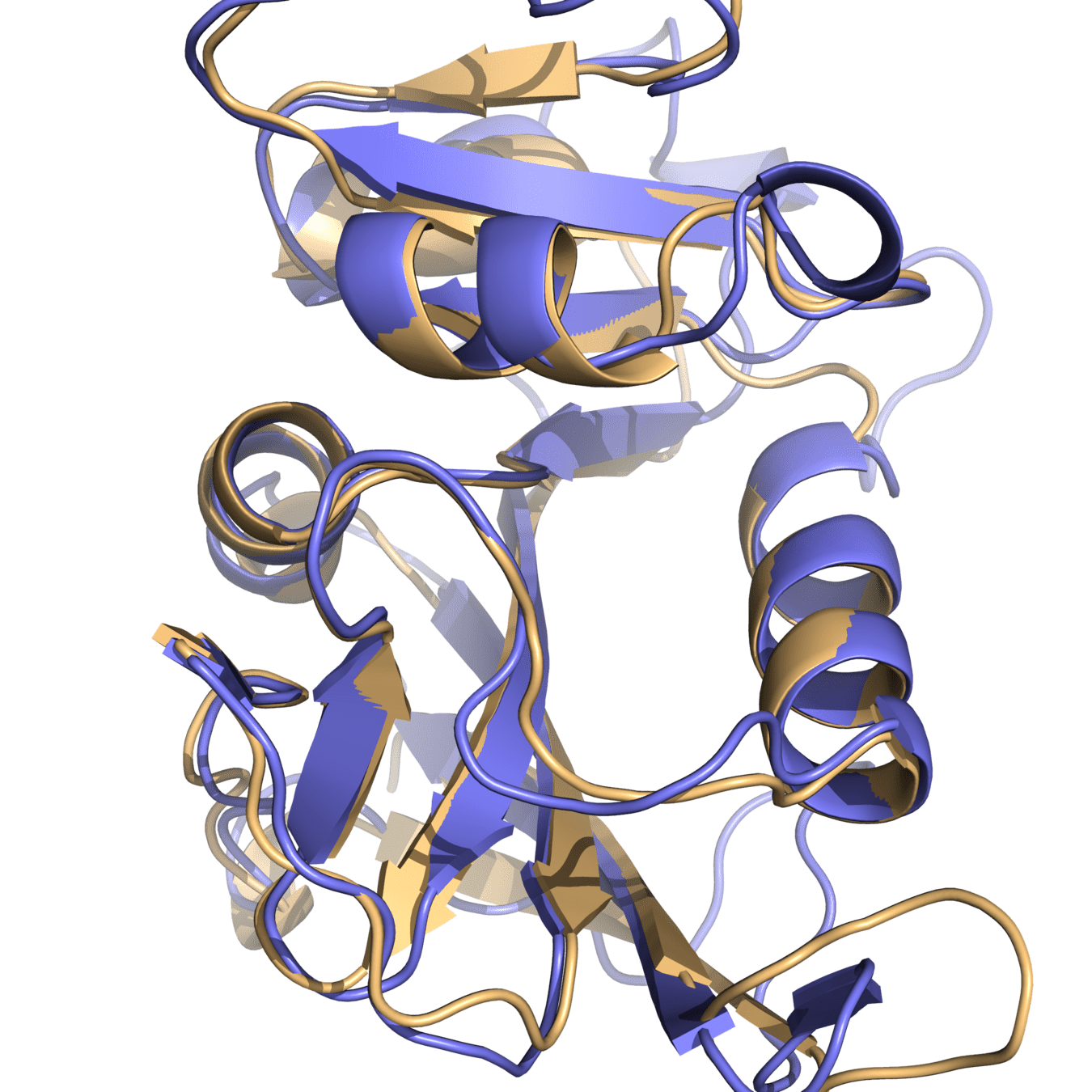
DHFR conservation complicates drug design
Bacteria and humans have similar structures, but their dynamics are different
Outcome: We need to ensure drugs only bind to bacterial proteins by exploiting dynamic insights
Simulating DHFR provides insight into druggable conformations
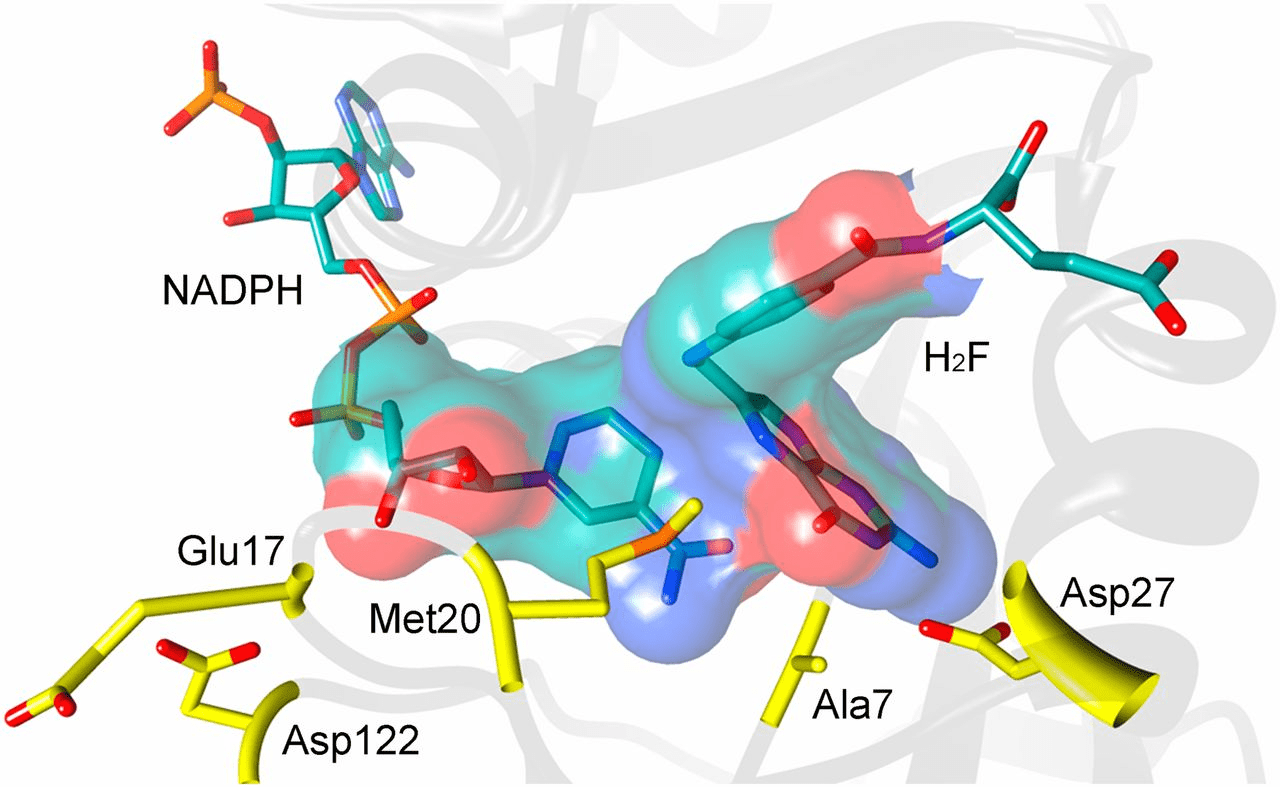
MD simulations will explore various low-energy conformations that are, hopefully, similar to reality
Knowing conformations unique to bacteria allow us to design a small molecule that competitively inhibits DHFR
After today, you should be able to
Select and prepare a protein structure for molecular simulations.
We need a structure before starting any molecular simulation
If our starting structure is very far away from our desired equilibrium, our simulations will take longer
For example, we would have to wait for the protein to fold to study it's dynamics
- Low-quality experimental structures
- Inaccurate computational predictions
- High-energy conformations
- Missing or incorrect cofactors
We can obtain starting structures from experimental databases
Experimental structures offer the best option for their accuracy
PDB contains experimentally determined structures for thousands of proteins
General resolution preference: X-ray, Cryo-EM, NMR
Not all structures in the PDB are equally suitable for simulations
Proteins can exist in different functional conformations: active vs. inactive state, bound to ligands or unbound
Functional state
Higher B-factors suggest more uncertainty in atom positions, which might make that part of the structure less reliable
B-factors
Flexible loops or disordered regions are often missing from the structure
Completeness
Resolution
The resolution of a structure refers to how well the atomic positions are determined
Tip: A resolution below 2.0 Å is generally preferred for high-quality simulations.
Not all structures in the PDB are equally suitable for simulations
| Factor | 7D4L | 4NX6 | 4KJK | 4NX7 |
|---|---|---|---|---|
| Resolution (Å) | 1.60 | 1.35 | 1.35 | 1.15 |
| Temperature | 298 | 298 | 298 | 100 |
| R-free | 0.196 | 0.190 | 0.166 | 0.170 |
| Clashscore | 2 | 5 | 8 | 12 |
| Ramachandran outliers | 0 | 0 | 0 | 0 |
| Rotamer outliers | 1 | 2 | 1 | 5 |
Here are some example structural characteristics with the best value in bold
7D4L is a good choice
Resolution and R-free are comparable, and few clashes are highly desirable
Reasonable structures will likely provide similar results
| Factor | 7D4L | 4KJK |
|---|---|---|
| Resolution | 1.60 | 1.35 |
| Temperature | 298 | 298 |
| R-free | 0.196 | 0.166 |
| Clashscore | 2 | 8 |
| Ramachandran outliers | 0 | 0 |
| Rotamer outliers | 1 | 1 |
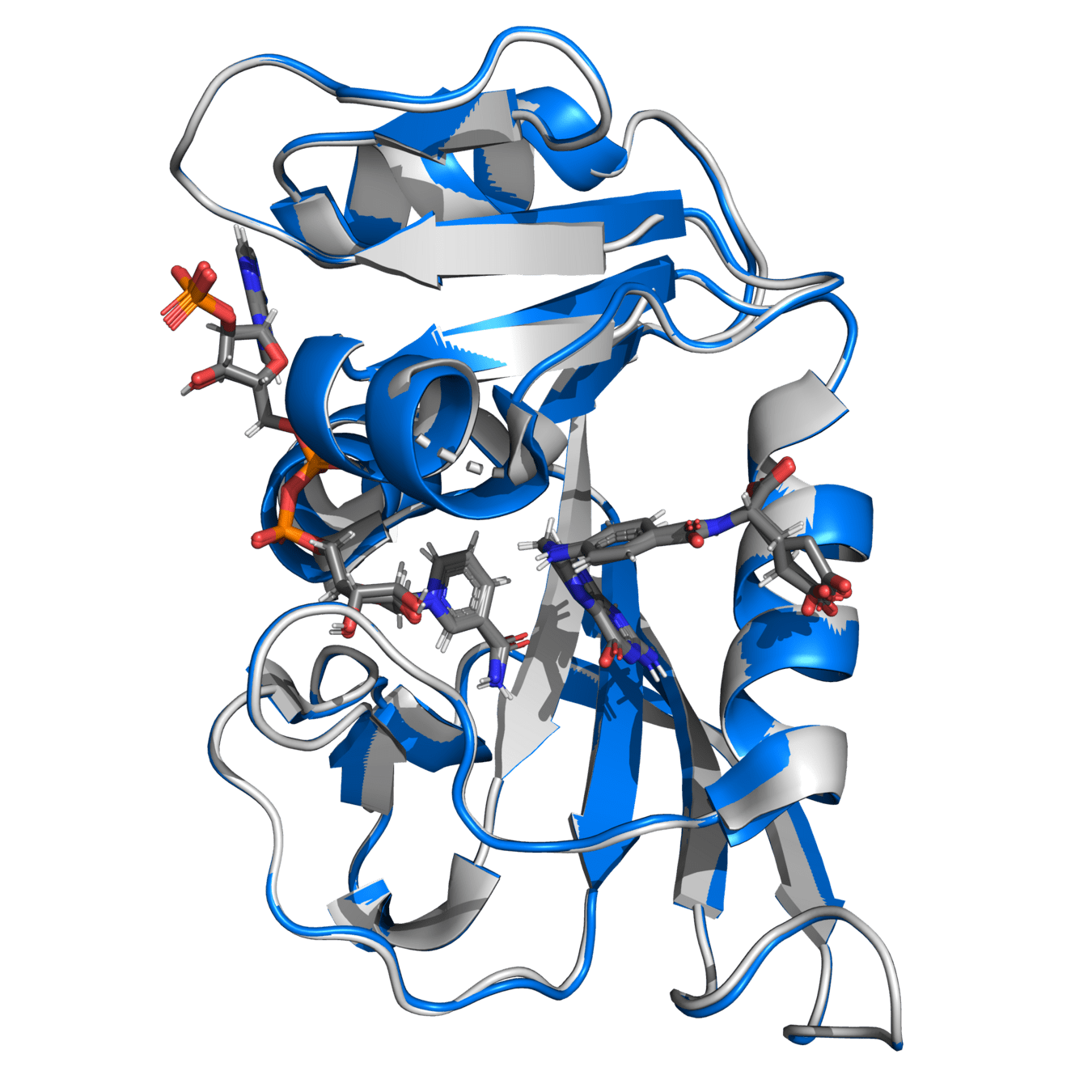
Alpha carbon RMSD is 0.141 (indicating high similarity)
Either structure would provide comparable results if simulation protocols are appropriate
Simulations cannot have missing residues
It’s essential to fix chain breaks and missing loops before simulation
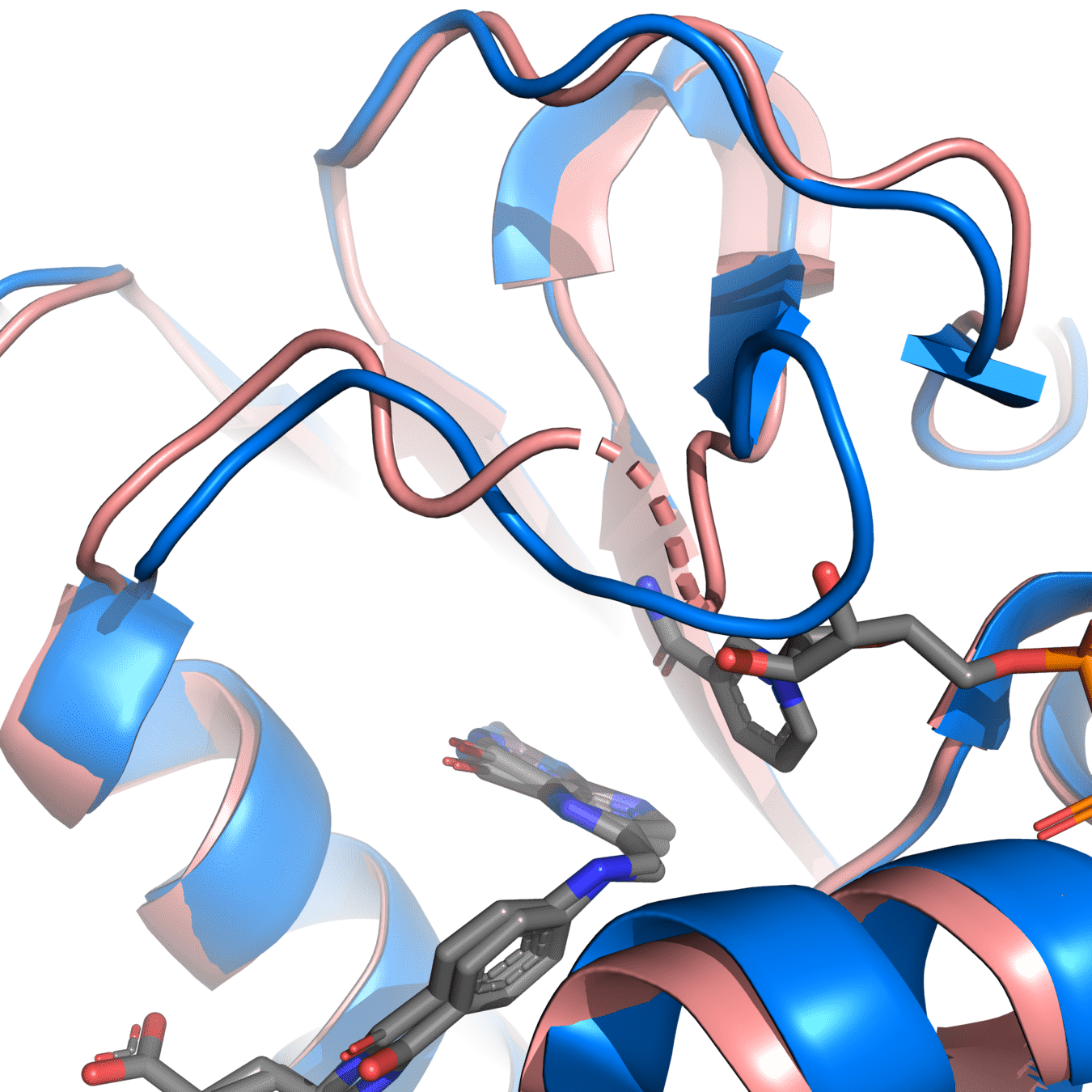
8UCX is missing residues 17 and 18
Protein structure predictions are used to add missing residues
Missing atoms or residues can be added using modeling software like Modeller
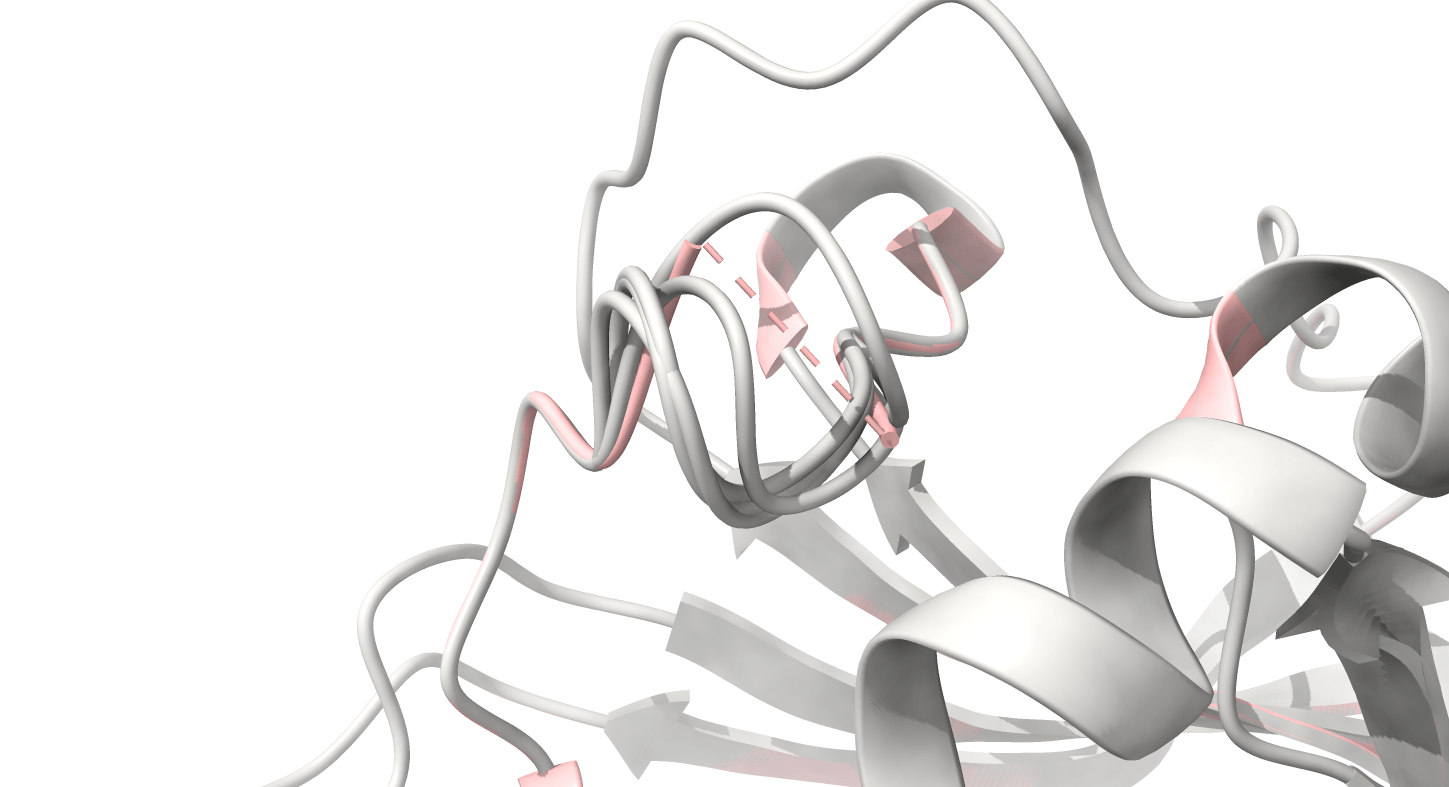
Pink: 8UCX
Grey: Modeller
Dashed lines often indicate missing atoms
Unwanted components like ligands or non-essential ions should be removed
Many PDB structures contain ligands, ions, or crystallization agents that are not physiologically relevant
These can distort the protein's behavior in a simulated biological environment if not removed
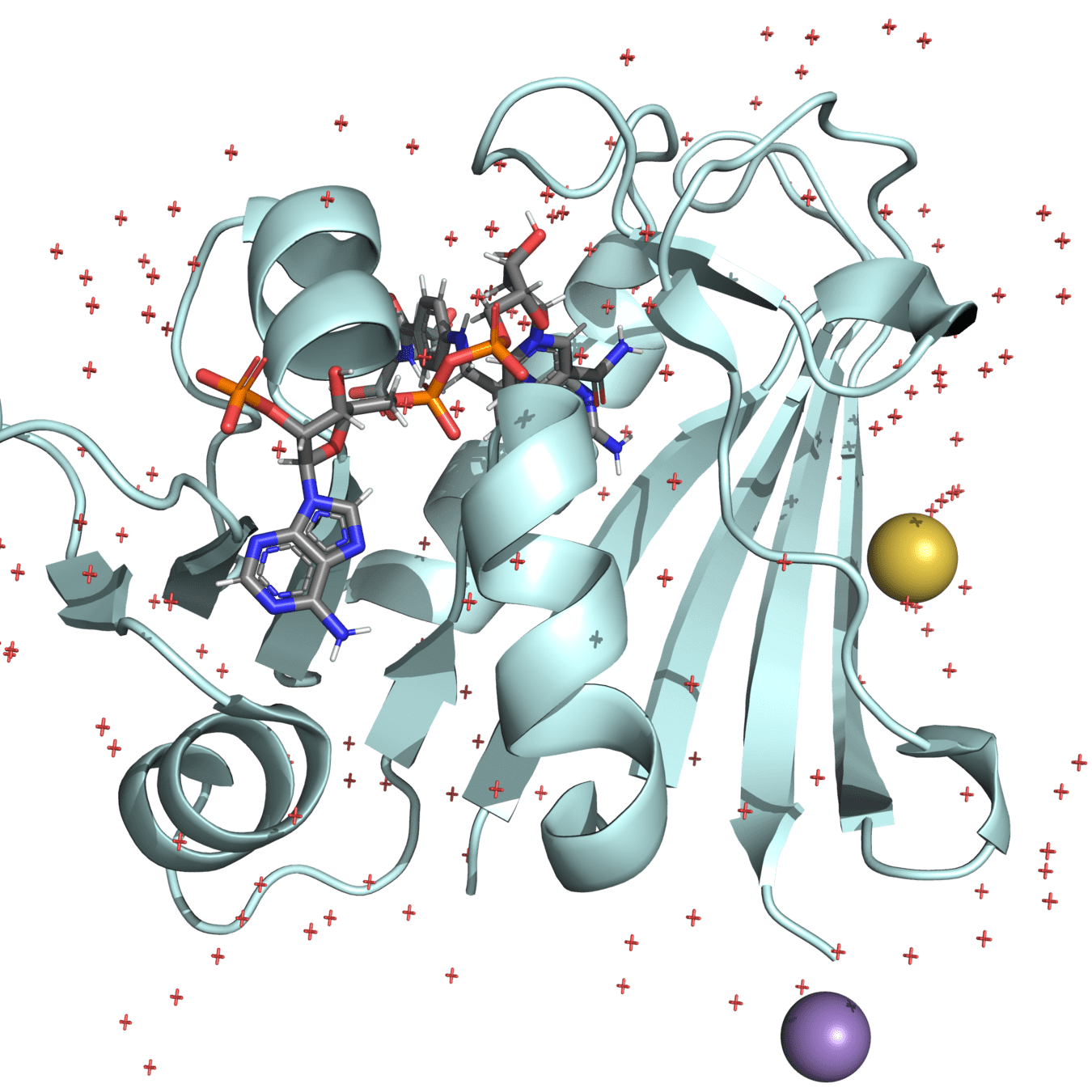
Manganese (II)
Mercaptoethanol
Water molecules
Ligands
Correct protonation states are essential for accurate simulations
Experimental structures often cannot resolve hydrogens, so we need to add them ourselves
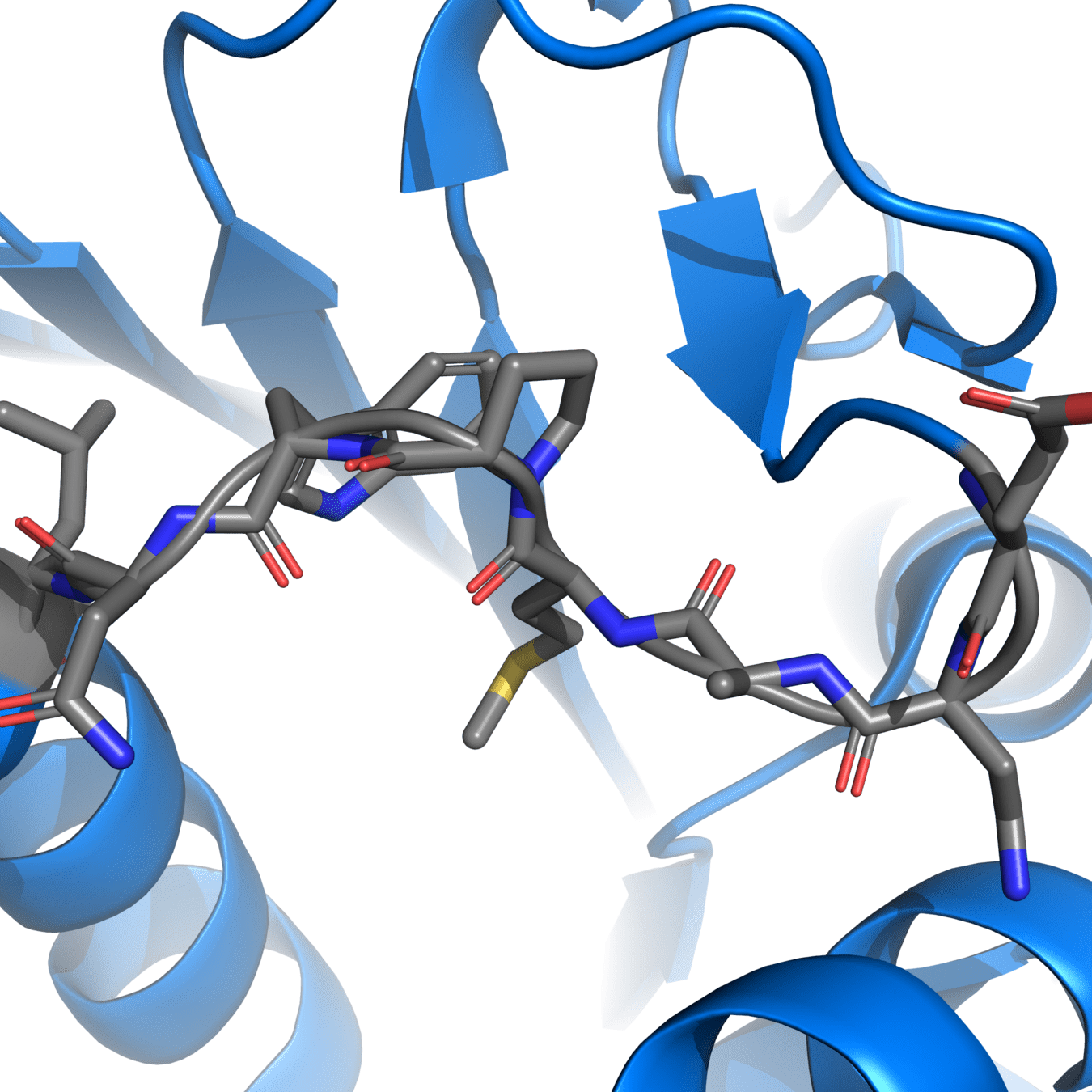
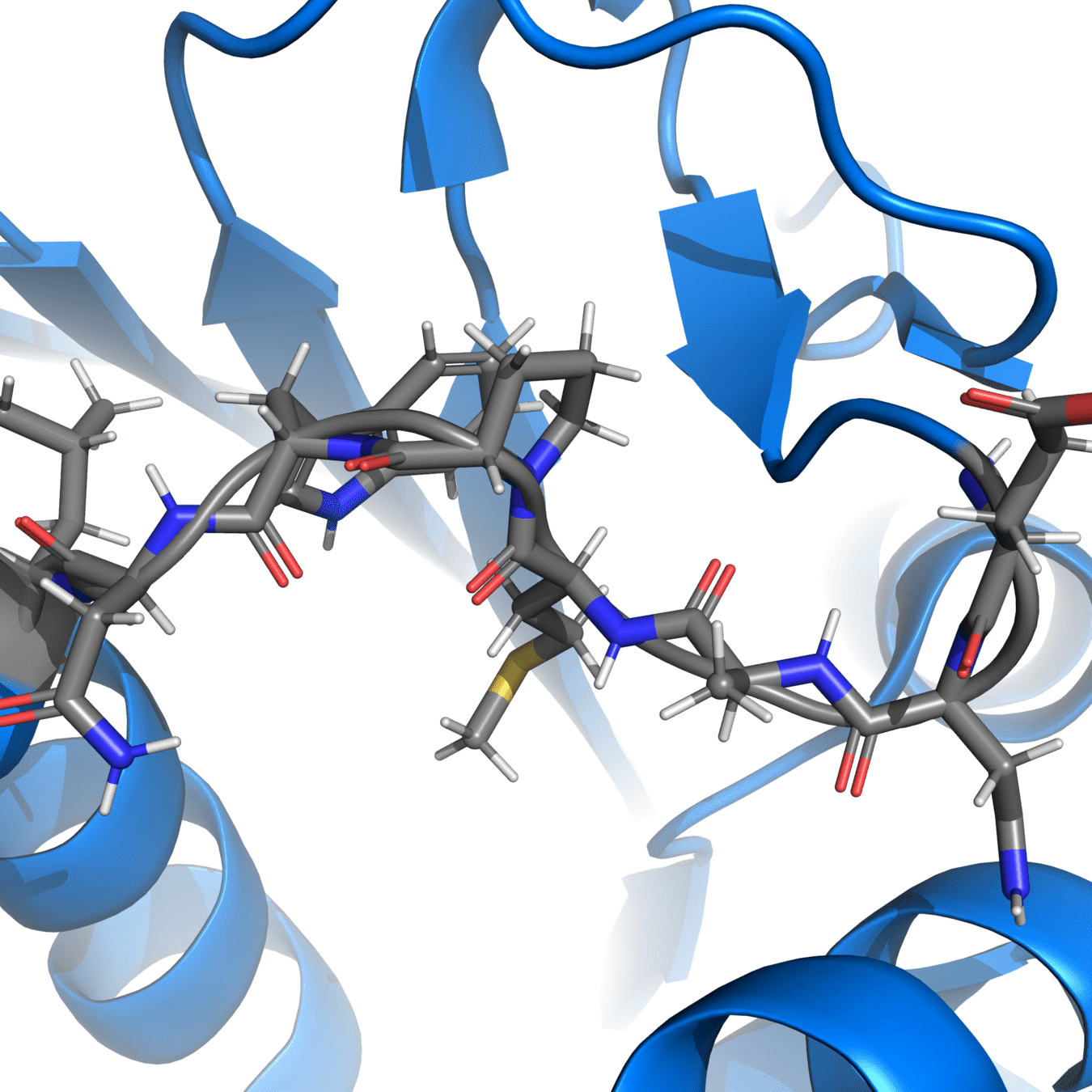
pH-sensitive residues
Histidine (His, H): pKa ~6.0
Protonation switching around pH 6 - 7
Cysteine (Cys, C): pKa ~8.3
Could form disulfide bonds in oxidizing environments
Aspartic Acid (Asp, D): pKa: ~3.9
Affects interactions like salt bridges and hydrogen bonds
Glutamic Acid (Glu, E): pKa: ~4.2
Glu's protonation state affects electrostatic interactions.
Lysine (Lys, K): pKa: ~10.5
Can form ionic bonds with negatively charged residues
Tyrosine (Tyr, Y): pKa: ~10.1
Hydrogen bonding and in enzyme active sites
Protonation states of amino acids affect the charge distribution, which influences electrostatic interactions during the simulation
We now have a fully prepared protein
After today, you should be able to
Explain the importance of approximating molecular environments.
DHFR is localized in the cytoplasm, which contains a multitude of chemical species
Ions
Potassium, Sodium, Calcium, Magnesium, Iron, Zinc, Copper, Manganese, Phosphate, Chloride, Bicarbonate, Sulfate, Citrate, ATP, ADP, AMP, . . .
Molecules
Glucose, pyruvate, lactate, amino acids, fatty acids, nucleotides, NADH, FADH, citrate, oxaloacetate, biotin, riboflavin, coenzyme A, ubiquinone, . . .
Proteins
Glycolytic enzymes, TCA cycle enzymes, DNA/RNA polymerases, kinases, phosphatases, G-proteins, heat shock proteins, molecular motors, transcription factors, transcription regulators, ribosomes, proteasomes, . . .
Organelles
Mitochondria, endoplasmic reticulum, golgi apparatus, lysosomes, peroxisomes, vacuoles, endosomes, ribosomes, centrosomes, . . .
Cytoskeleton
Actin, profilin, cofilin, myosin, keratins, vimentin, neurofilaments, tubulin, . . .
Membranes
Phospholipid bilayer with embedded proteins, cholesterol, clycoproteins, glycolipids, . . .
and more

Simulations should accurately represent reality
What biological or chemical components are crucial for modeling the dynamics of a protein in the cytosol?

We must balance computational feasibility with biological realism
- Protein of interest (already prepared)
- Water molecular at the appropriate temperature (310 K) and pressure (1 atm)
- Cations (Na+ or K+) and anions (Cl-) at an ionic strength of 150 millimolar
- Any cofactors (e.g., NADPH and Folate for DHFR)
Example of system: roGPF2
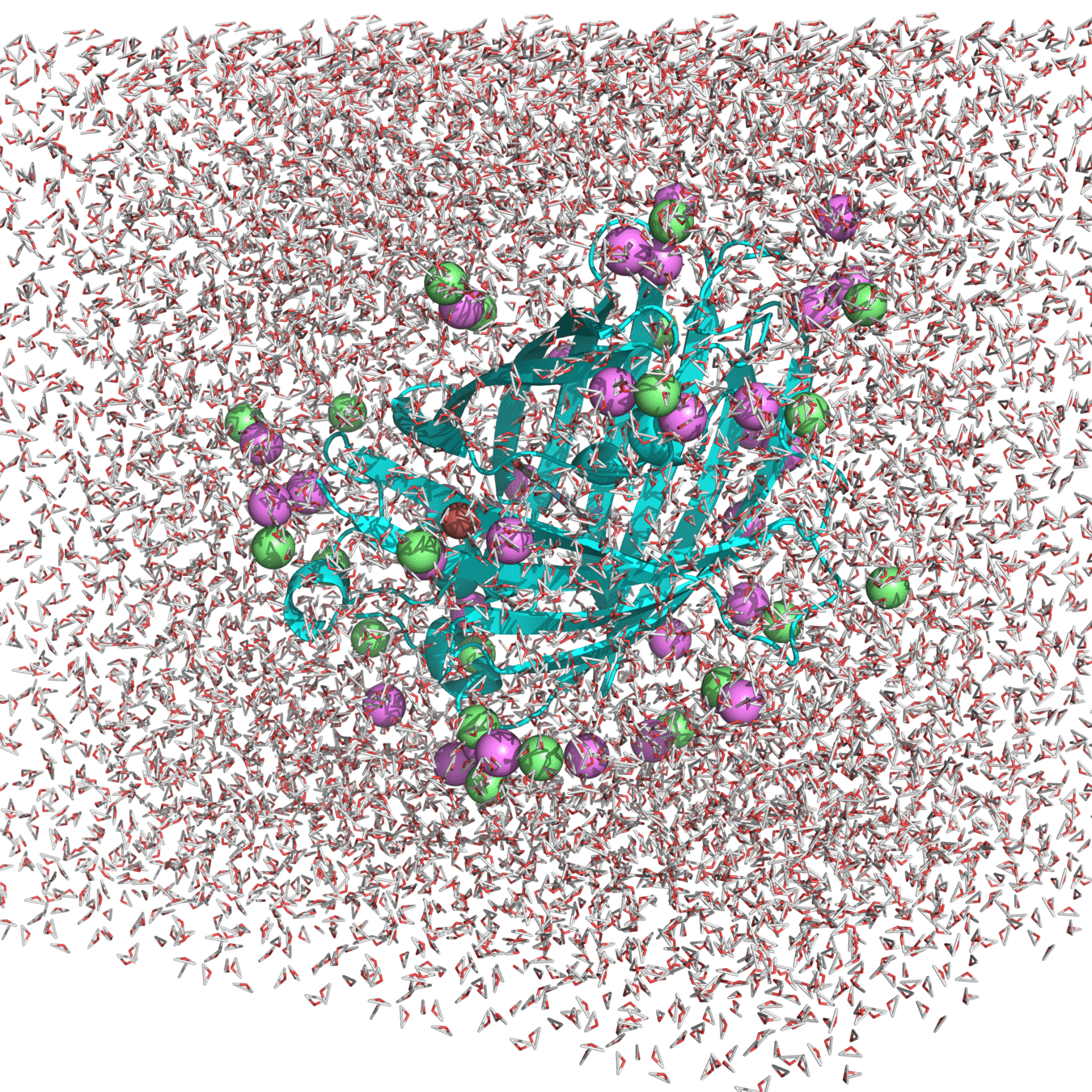
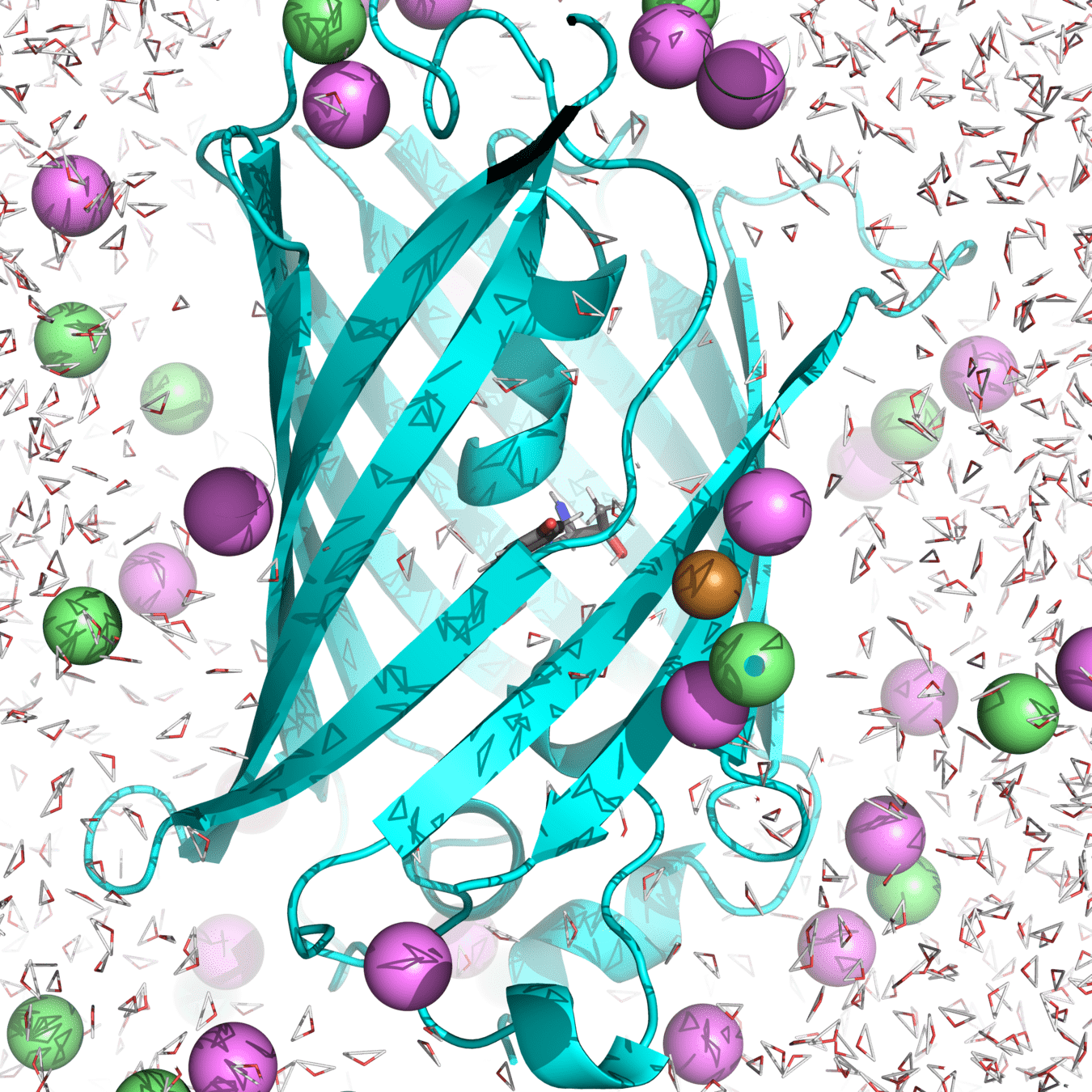
Starting structure for simulating Cu(I) binding to Cys147 and 204 in roGFP2 with Na+ and Cl- counterions
(Actually used in my research.)
After today, you should be able to
Describe periodic boundary conditions and their role in MD simulations.
Realistic systems do not have walls
For this simulation, we would have to apply a force to keep the molecules in this box
Water molecules and proteins would bounce off these walls in an unphysical manner (i.e., edge effects)
A protein in vivo or in vitro will have plenty of space to move around
We could make the box very large, but this would dramatically increase the cost
Periodic boundary conditions (PBC) is how we solve this issue
PBC simulate infinite systems from a finite box
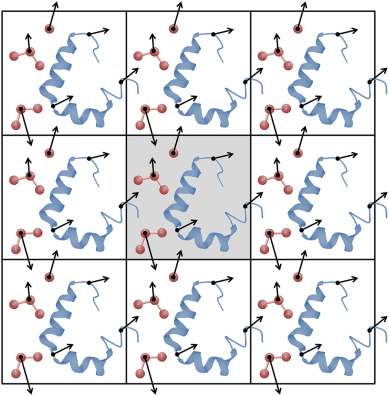
Think PackMan: If he crosses the right side of the map, he reappears on the left

We (virually) place exact copies of our system in all directions
Atoms that cross the box edge reappear on the other side; thus, do not have edge effects
The minimum image convention ensure correct interaction
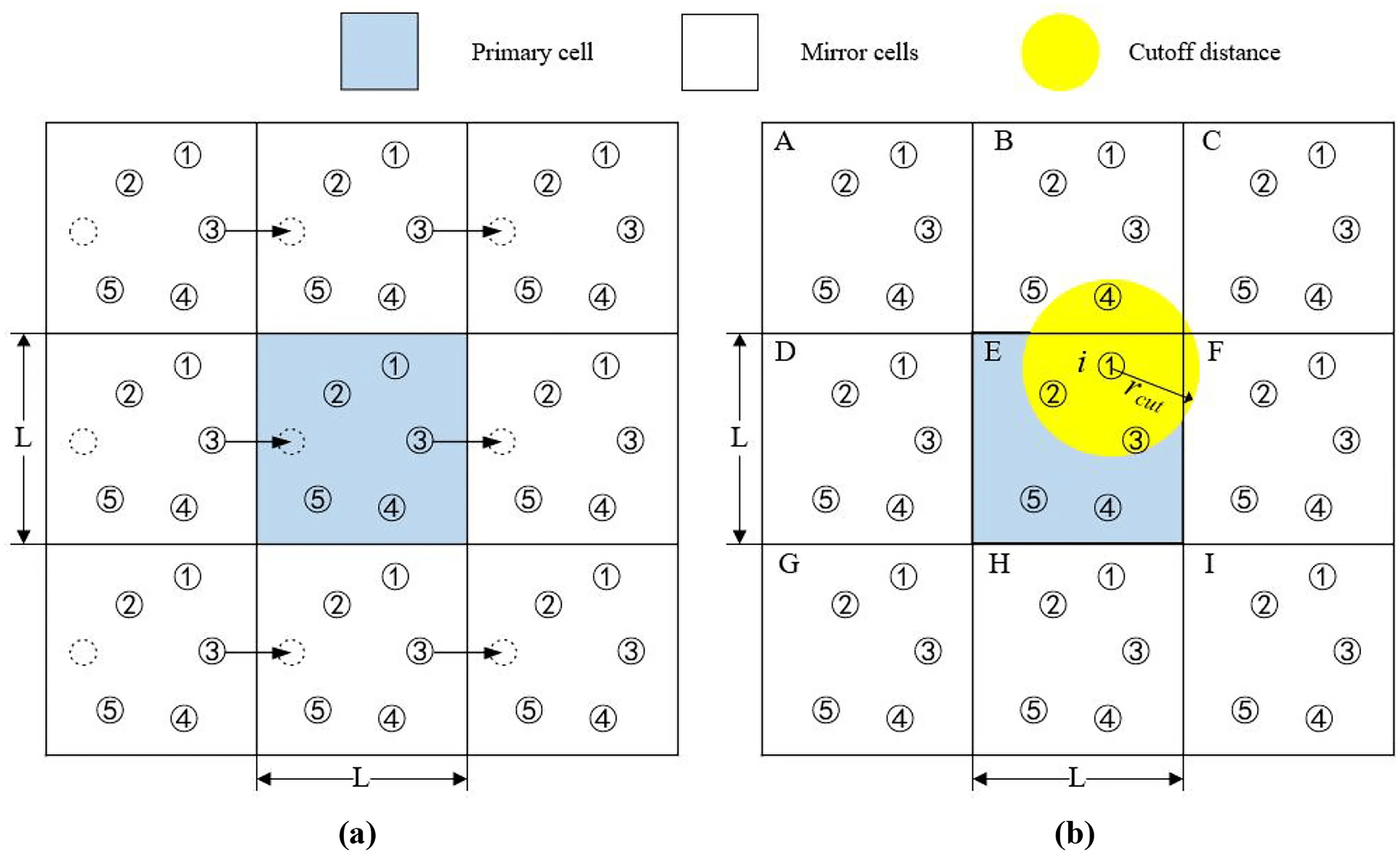
Image atoms in adjacent boxes are used to calculate interactions across the boundaries
The minimum image convention (MIC) ensures that an atom in the primary box only interacts with the closest image of another atom
After today, you should be able to
Explain the role of force field selection and topology generation.
We now have a fully prepared system, now we prepare our simulation
Force fields are parameterized to reproduce quantum chemical and experimental data
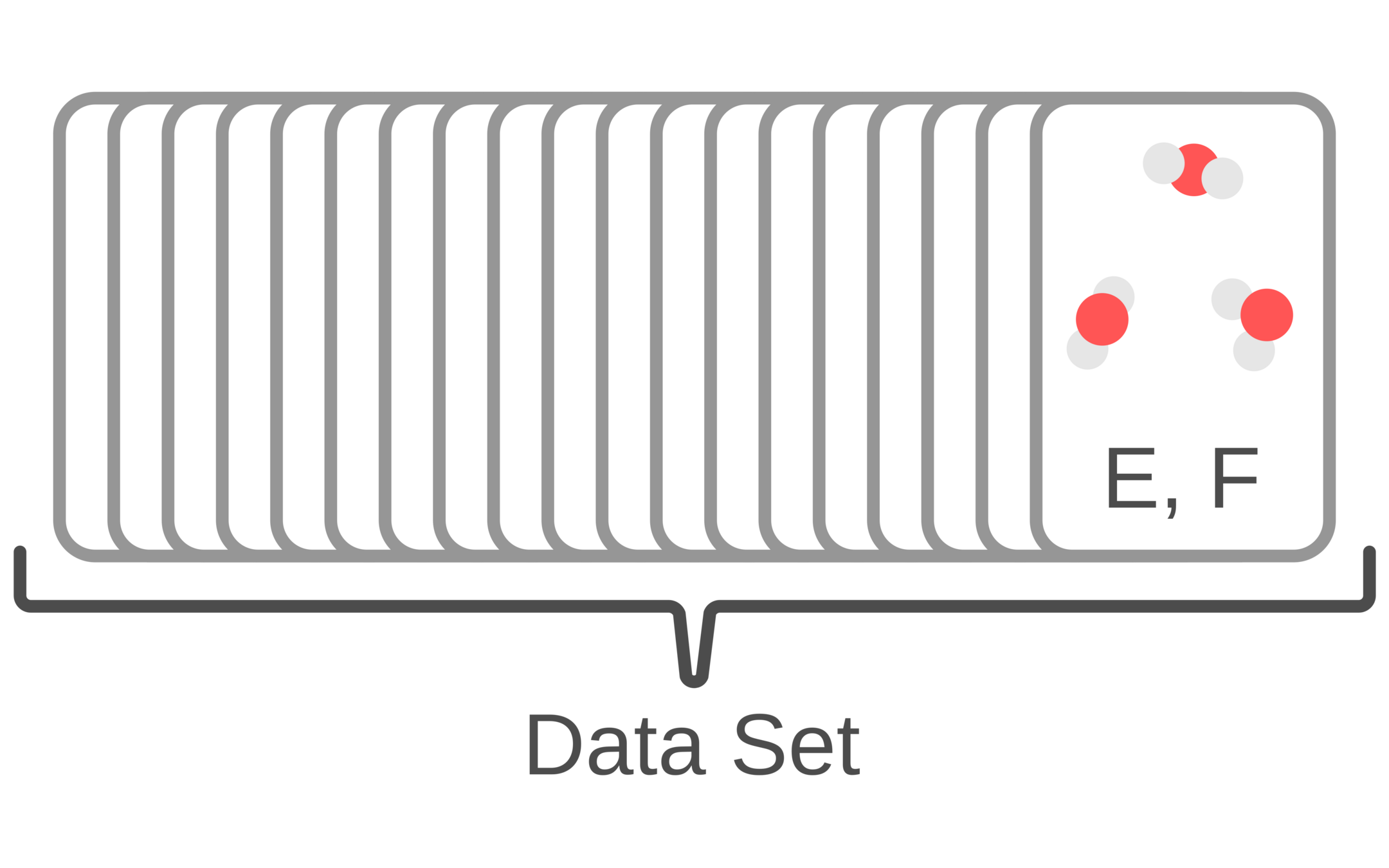
1. Generate structures and use quantum chemistry to compute energy and forces
2. Optimize force field parameters until they reproduce the quantum chemistry dataset
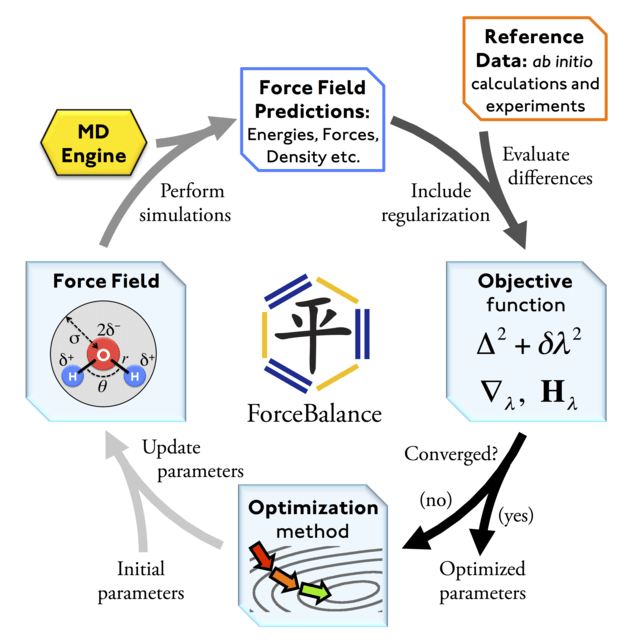
3. Run MD simulations and predict experimental data (e.g., NMR, Raman spectroscopy, solvation energies, etc.)
4. Continue to optimize force field parameters to minimizing quantum chemistry and simulation prediction errors
Force fields are dependent on fitting data and simulation setup
Force fields are not inherently compatible with each other

Example: Simulating a DNA-binding protein
Suppose my protein force field was fit to:
- Membrane proteins
- Proteins and RNA
Suppose my DNA force field was fit to:
- Single-stranded DNA
- Protein binding with a different type of force field
Simulations would be unreliable because the force fields are incompatible with each other
Forcefields are compatible by design, or are validated against experimental data
Key factors for selecting a force field
- System type: Different force fields are optimized for specific systems
- Accuracy vs. speed: High-accuracy force fields may require more computational resources
- Compatibility: Choose a force field based on compatibility with available topology generators and the type of molecules in your simulation.
- AMBER: Best for proteins and nucleic acids, optimized for biomolecular interactions.
- CHARMM: Known for its extensive parameter set, suitable for complex systems including proteins, lipids, and membranes.
- OPLS: Optimized for small molecules, organic compounds, and polymers, with emphasis on accurate non-bonded interactions.
Examples:
Topology files define the molecular structure and interactions in a simulation
A topology file contains information on atom types, bonds, angles, dihedrals, and non-bonded interactions based on the chosen force field

Essentially tells the program which force field parameters to use where
Example AMBER topology
%VERSION VERSION_STAMP = V0001.000 DATE = 01/20/24 21:37:10
%FLAG TITLE
%FORMAT(20a4)
default_name
%FLAG POINTERS
%FORMAT(10I8)
33582 19 31714 1852 4022 2505 8139 7890 0 0
59724 10270 1852 2505 7890 89 205 205 47 0
0 0 0 0 0 0 0 1 36 0
0
%FLAG ATOM_NAME
%FORMAT(20a4)
N H1 H2 H3 CA HA CB HB2 HB3 CG HG2 HG3 SD CE HE1 HE2 HE3 C O N
H CA HA CB HB2 HB3 OG HG C O N H CA HA CB HB2 HB3 CG HG2 HG3
CD HD2 HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C O N H CA HA2 HA3 C O N
H CA HA CB HB2 HB3 CG HG2 HG3 CD OE1 OE2 C O N H CA HA CB HB2
HB3 CG HG2 HG3 CD OE1 OE2 C O N H CA HA CB HB2 HB3 CG HG CD1 HD11
HD12HD13CD2 HD21HD22HD23C O N H CA HA CB HB2 HB3 CG CD1 HD1 CE1 HE1
CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1
C O N H CA HA2 HA3 C O N H CA HA CB HB CG1 HG11HG12HG13CG2
HG21HG22HG23C O N H CA HA CB HB CG1 HG11HG12HG13CG2 HG21HG22HG23C
O N CD HD2 HD3 CG HG2 HG3 CB HB2 HB3 CA HA C O N H CA HA CB
HB CG2 HG21HG22HG23CG1 HG12HG13CD1 HD11HD12HD13C O N H CA HA CB HB2
HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22HD23C O N H CA HA CB HB CG1
HG11HG12HG13CG2 HG21HG22HG23C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD
OE1 OE2 C O N H CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22
HD23C O N H CA HA CB HB2 HB3 CG OD1 OD2 C O N H CA HA2 HA3
C O N H CA HA CB HB2 HB3 CG OD1 OD2 C O N H CA HA CB HB
CG1 HG11HG12HG13CG2 HG21HG22HG23C O N H CA HA CB HB2 HB3 CG OD1 ND2
HD21HD22C O N H CA HA2 HA3 C O N H CA HA CB HB2 HB3 CG ND1
HD1 CE1 HE1 NE2 CD2 HD2 C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2
HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C O N H CA HA CB HB2 HB3 CG CD1 HD1
CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA CB HB2 HB3 OG HG C
O N H CA HA CB HB CG1 HG11HG12HG13CG2 HG21HG22HG23C O N H CA
HA CB HB2 HB3 OG HG C O N H CA HA2 HA3 C O N H CA HA CB
HB2 HB3 CG HG2 HG3 CD OE1 OE2 C O N H CA HA2 HA3 C O N H CA
HA CB HB2 HB3 CG HG2 HG3 CD OE1 OE2 C O N H CA HA2 HA3 C O N
H CA HA CB HB2 HB3 CG OD1 OD2 C O N H CA HA CB HB1 HB2 HB3 C
O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C O N H CA HA CB
HB2 HB3 CG CD1 HD1 CE1 HE1 CZ OH HH CE2 HE2 CD2 HD2 C O N H CA HA2
HA3 C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3 NZ
HZ1 HZ2 HZ3 C O N H CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21
HD22HD23C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C O N H
CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22HD23C O N H CA
HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C O N
H CA HA CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N
H CA HA CB HB CG2 HG21HG22HG23CG1 HG12HG13CD1 HD11HD12HD13C O N H
CA HA CB HB2 HB3 OG HG C O N H CA HA CB HB CG2 HG21HG22HG23OG1
HG1 C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C O N H CA
HA2 HA3 C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3
NZ HZ1 HZ2 HZ3 C O N H CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2
HD21HD22HD23C O N CD HD2 HD3 CG HG2 HG3 CB HB2 HB3 CA HA C O N
H CA HA CB HB CG1 HG11HG12HG13CG2 HG21HG22HG23C O N CD HD2 HD3 CG
HG2 HG3 CB HB2 HB3 CA HA C O N H CA HA CB HB2 HB3 CG CD1 HD1 NE1
HE1 CE2 CZ2 HZ2 CH2 HH2 CZ3 HZ3 CE3 HE3 CD2 C O N CD HD2 HD3 CG HG2 HG3
CB HB2 HB3 CA HA C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C
O N H CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22HD23C O
N H CA HA CB HB CG1 HG11HG12HG13CG2 HG21HG22HG23C O N H CA HA
CB HB CG2 HG21HG22HG23OG1 HG1 C O N H CA HA CB HB CG2 HG21HG22HG23
OG1 HG1 C O N H CA HA CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22
HD23C O CD2 CE2 CZ CG2 CD1 CE1 CB2 CA2 C2 H10 H12 H11 H9 H8 OH O2 N2
N3 C1 CA3 CA1 H13 H14 C3 O3 N1 H1 H2 CB1 CG1 H5 H6 H7 H3 OG1 H4 N
H CA HA CB HB CG1 HG11HG12HG13CG2 HG21HG22HG23C O N H CA HA CB
HB2 HB3 CG HG2 HG3 CD OE1 NE2 HE21HE22C O N H CA HA CB HB2 HB3 SG
HG C O N H CA HA CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2
HD2 C O N H CA HA CB HB2 HB3 OG HG C O N H CA HA CB HB2
HB3 CG HG2 HG3 CD HD2 HD3 NE HE CZ NH1 HH11HH12NH2 HH21HH22C O N H
CA HA CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ OH HH CE2 HE2 CD2 HD2 C O N
CD HD2 HD3 CG HG2 HG3 CB HB2 HB3 CA HA C O N H CA HA CB HB2 HB3
CG OD1 OD2 C O N H CA HA CB HB2 HB3 CG ND1 HD1 CE1 HE1 NE2 CD2 HD2
C O N H CA HA CB HB2 HB3 CG HG2 HG3 SD CE HE1 HE2 HE3 C O N
H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C
O N H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 NE HE CZ NH1 HH11HH12
NH2 HH21HH22C O N H CA HA CB HB2 HB3 CG ND1 CE1 HE1 NE2 HE2 CD2 HD2
C O N H CA HA CB HB2 HB3 CG OD1 OD2 C O N H CA HA CB HB2
HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA CB HB2
HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA CB HB2
HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C O N H CA HA
CB HB2 HB3 OG HG C O N H CA HA CB HB1 HB2 HB3 C O N H CA
HA CB HB2 HB3 CG HG2 HG3 SD CE HE1 HE2 HE3 C O N CD HD2 HD3 CG HG2
HG3 CB HB2 HB3 CA HA C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD OE1
OE2 C O N H CA HA2 HA3 C O N H CA HA CB HB2 HB3 CG CD1 HD1
CE1 HE1 CZ OH HH CE2 HE2 CD2 HD2 C O N H CA HA CB HB CG1 HG11HG12
HG13CG2 HG21HG22HG23C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD OE1 NE2
HE21HE22C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD OE1 OE2 C O N
H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 NE HE CZ NH1 HH11HH12NH2 HH21
HH22C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C O N H CA
HA CB HB CG2 HG21HG22HG23CG1 HG12HG13CD1 HD11HD12HD13C O N H CA HA
CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA
CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ HZ CE2 HE2 CD2 HD2 C O N H CA HA
CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3 NZ HZ1 HZ2 HZ3 C O N H
CA HA CB HB2 HB3 CG OD1 OD2 C O N H CA HA CB HB2 HB3 CG OD1 OD2
C O N H CA HA2 HA3 C O N H CA HA CB HB2 HB3 CG OD1 ND2 HD21
HD22C O N H CA HA CB HB2 HB3 CG CD1 HD1 CE1 HE1 CZ OH HH CE2 HE2
CD2 HD2 C O N H CA HA CB HB2 HB3 CG HG2 HG3 CD HD2 HD3 CE HE2 HE3
NZ HZ1 HZ2 HZ3 C O N H CA HA CB HB CG2 HG21HG22HG23OG1 HG1 C O We never actually look at these files
Complex molecules and ligands requires parameterization and careful integration
Non-standard residues or ligands are not always included in standard force field parameter sets
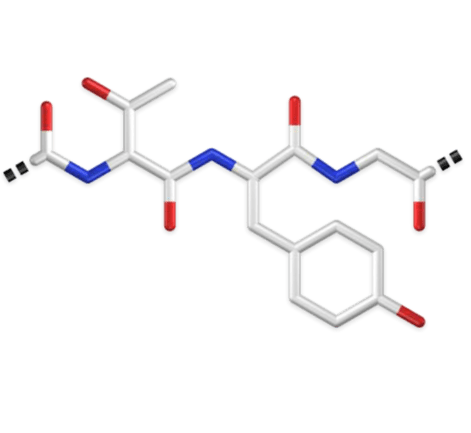
These require additional parameterization to ensure proper interactions in the simulation
Example: GFP chromophore
After today, you should be able to
Outline the process of energy minimization and its significance.
Energy minimization is necessary before running molecular dynamics simulations
Energy minimization adjusts the initial structure to remove unfavorable atom positions and steric clashes that could cause instability during simulations
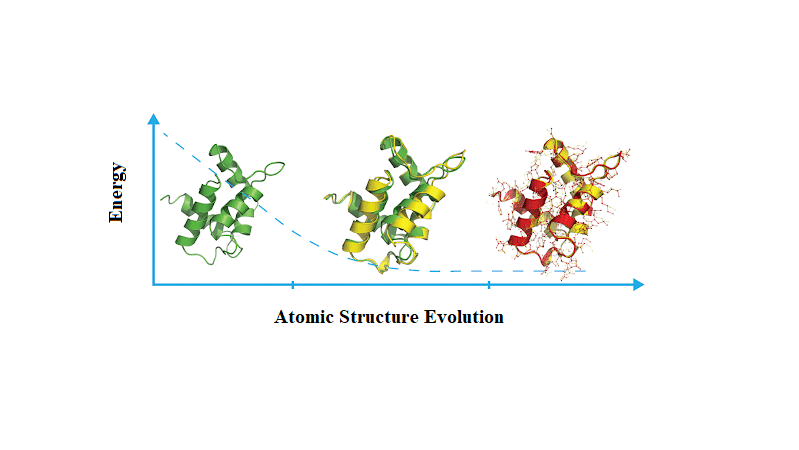
Without minimization, high-energy configurations may lead to unrealistic results or early failures in the molecular dynamics simulation
Energy minimization removes steric clashes and optimizes the initial geometry
Steric clashes occur when atoms are too close together, resulting in excessively high energy
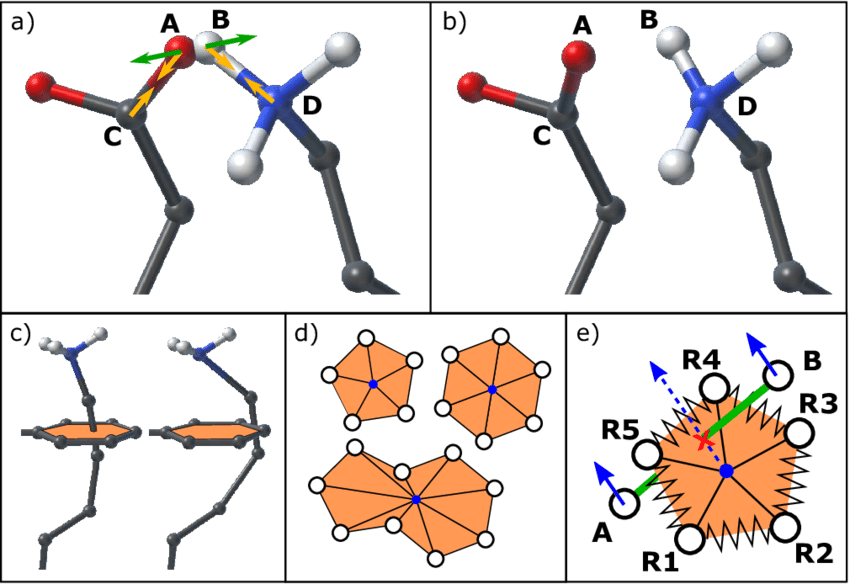
Energy minimization gently adjusts the structure to lower the system’s energy
Unphysical
Physical
Before the next class, you should
- Work on A05
Lecture 14:
Molecular system representations
Today
Thursday
Lecture 15:
Atomistic insights
BIOSC 1540: L14 (Molecular system representations)
By aalexmmaldonado



