Generic solutions for specific problems: Snakemake, Datavzrd, Bioconda,
and Varlociraptor in 2024
Johannes Köster
GCB 2024


Snakemake
Snakemake
>1 million downloads since 2015
>3000 citations
>12 citations per week in 2023
affiliated


Mölder, F., Jablonski, K.P., Letcher, B., Hall, M.B., Tomkins-Tinch, C.H., Sochat, V., Forster, J., Lee, S., Twardziok, S.O., Kanitz, A., Wilm, A., Holtgrewe, M., Rahmann, S., Nahnsen, S., Köster, J. Sustainable data analysis with Snakemake. F1000Res 10, 33 (2021).
https://snakemake.github.io
dataset
results
dataset
dataset
dataset
dataset
dataset
https://snakemake.github.io
Define workflows
in terms of rules
https://snakemake.github.io
Define workflows
in terms of rules
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
script:
"scripts/myscript.R"
rule myfiltration:
input:
"result/{dataset}.txt"
output:
"result/{dataset}.filtered.txt"
shell:
"mycommand {input} > {output}"
rule aggregate:
input:
"results/dataset1.filtered.txt",
"results/dataset2.filtered.txt"
output:
"plots/myplot.pdf"
script:
"scripts/myplot.R"https://snakemake.github.io
Define workflows
in terms of rules
Define workflows
in terms of rules
rule mytask:
input:
"data/{sample}.txt"
output:
"result/{sample}.txt"
params:
someparam=0.1
conda:
"envs/sometool.yaml"
shell:
"some-tool {params.someparam} {input} > {output}"- simple paths
- helpers for branching
and aggregation - Python logic
- shell commands
- ad-hoc scripts and notebooks (Python, R, Julia, Rust, Bash)
- wrappers
https://snakemake.github.io
deployment via
conda, containers
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
script:
"scripts/myscript.R"
rule myfiltration:
input:
"result/{dataset}.txt"
output:
"result/{dataset}.filtered.txt"
shell:
"mycommand {input} > {output}"
rule aggregate:
input:
"results/dataset1.filtered.txt",
"results/dataset2.filtered.txt"
output:
"plots/myplot.pdf"
script:
"scripts/myplot.R"Automatic inference of DAG of jobs

https://snakemake.github.io
Scalable to any platform



workstation
compute server
cluster
grid computing
cloud computing

https://snakemake.github.io
Self-contained HTML reports
https://snakemake.github.io
Snakemake 8: plugin architecture
https://snakemake.github.io

Simplicity and automation
Object oriented implementation of a few abstract methods
class Executor(RemoteExecutor):
def run_job(self, job: JobExecutorInterface):
...
async def check_active_jobs(
self, active_jobs: List[SubmittedJobInfo]
) -> Generator[SubmittedJobInfo, None, None]:
...
def cancel_jobs(self, active_jobs: List[SubmittedJobInfo]):
...
https://snakemake.github.io
Simplicity and automation
Dataclass for CLI arg registry
@dataclass
class ExecutorSettings(ExecutorSettingsBase):
namespace: str = field(
default="default", metadata={"help": "The namespace to use for submitted jobs."}
)
cpu_scalar: float = field(
default=0.95,
metadata={
"help": "K8s reserves some proportion of available CPUs for its own use. "
"So, where an underlying node may have 8 CPUs, only e.g. 7600 milliCPUs "
"are allocatable to k8s pods (i.e. snakemake jobs). As 8 > 7.6, k8s can't "
"find a node with enough CPU resource to run such jobs. This argument acts "
"as a global scalar on each job's CPU request, so that e.g. a job whose "
"rule definition asks for 8 CPUs will request 7600m CPUs from k8s, "
"allowing it to utilise one entire node. N.B: the job itself would still "
"see the original value, i.e. as the value substituted in {threads}."
},
)
...https://snakemake.github.io
Simplicity and automation
Poetry template for skeleton code
# Install poetry plugin via
poetry self add poetry-snakemake-plugin
# Create a new poetry project via
poetry new snakemake-executor-plugin-myfancyexecutor
cd snakemake-executor-plugin-myfancyexecutor
# Scaffold the project as a snakemake executor plugin
poetry scaffold-snakemake-executor-pluginhttps://snakemake.github.io
Simplicity and automation
Unified test suites for each plugin type
class TestWorkflows(snakemake.common.tests.TestWorkflowsLocalStorageBase):
__test__ = True
def get_executor(self) -> str:
return "slurm"
def get_executor_settings(self) -> Optional[ExecutorSettingsBase]:
return ExecutorSettings()
class TestWorkflowsRequeue(TestWorkflows):
def get_executor_settings(self) -> Optional[ExecutorSettingsBase]:
return ExecutorSettings(requeue=True)https://snakemake.github.io
Simplicity and automation
Automatic documentation
https://snakemake.github.io
Bioconda
Software installation is heterogeneous
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2")easy_install snakemake./configure --prefix=/usr/local
make
make installcp lib/amd64/jli/*.so lib
cp lib/amd64/*.so lib
cp * $PREFIXcpan -i bioperlcmake ../../my_project \
-DCMAKE_MODULE_PATH=~/devel/seqan/util/cmake \
-DSEQAN_INCLUDE_PATH=~/devel/seqan/include
make
make installapt-get install bwayum install python-h5pyinstall.packages("matrixpls")https://bioconda.github.io
Package management with Conda
package:
name: seqtk
version: 1.2
source:
fn: v1.2.tar.gz
url: https://github.com/lh3/seqtk/archive/v1.2.tar.gz
requirements:
build:
- gcc
- zlib
run:
- zlib
about:
home: https://github.com/lh3/seqtk
license: MIT License
summary: Seqtk is a fast and lightweight tool for processing sequences
test:
commands:
- seqtk seq#!/bin/bash
export C_INCLUDE_PATH=${PREFIX}/include
export LIBRARY_PATH=${PREFIX}/lib
make all
mkdir -p $PREFIX/bin
cp seqtk $PREFIX/bin- source or binary
- recipe and build script
- package
- install into isolated environments without admin permissions
https://bioconda.github.io
The Bioconda Project
B Grüning, R Dale, A Sjödin, BA Chapman, J Rowe, CH Tomkins-Tinch, R Valieris, ..., J Köster.
Bioconda: Sustainable and Comprehensive Software Distribution for the Life Sciences. Nature Methods 2018.
https://bioconda.github.io

a Conda channel for the life sciences

The Bioconda Project
B Grüning, R Dale, A Sjödin, BA Chapman, J Rowe, CH Tomkins-Tinch, R Valieris, ..., J Köster.
Bioconda: Sustainable and Comprehensive Software Distribution for the Life Sciences. Nature Methods 2018.
https://bioconda.github.io
>52000
commits
>48000
pull requests
>10800
packages
ISMB Proceedings 2023


Software
Scripts
https://bioconda.github.io
Recently added:
ARM support (Apple Silicon and Linux)
Potentially up next:
WASM support
https://bioconda.github.io
Datavzrd
Tables are the central entity
in data analysis

https://datavzrd.github.io
Not always a single table
oncoprint + individual variant calls
differentially expressed genes + expression matrix
https://datavzrd.github.io
Not always just a table
oncoprint + individual variant calls +
differentially expressed genes + expression matrix +
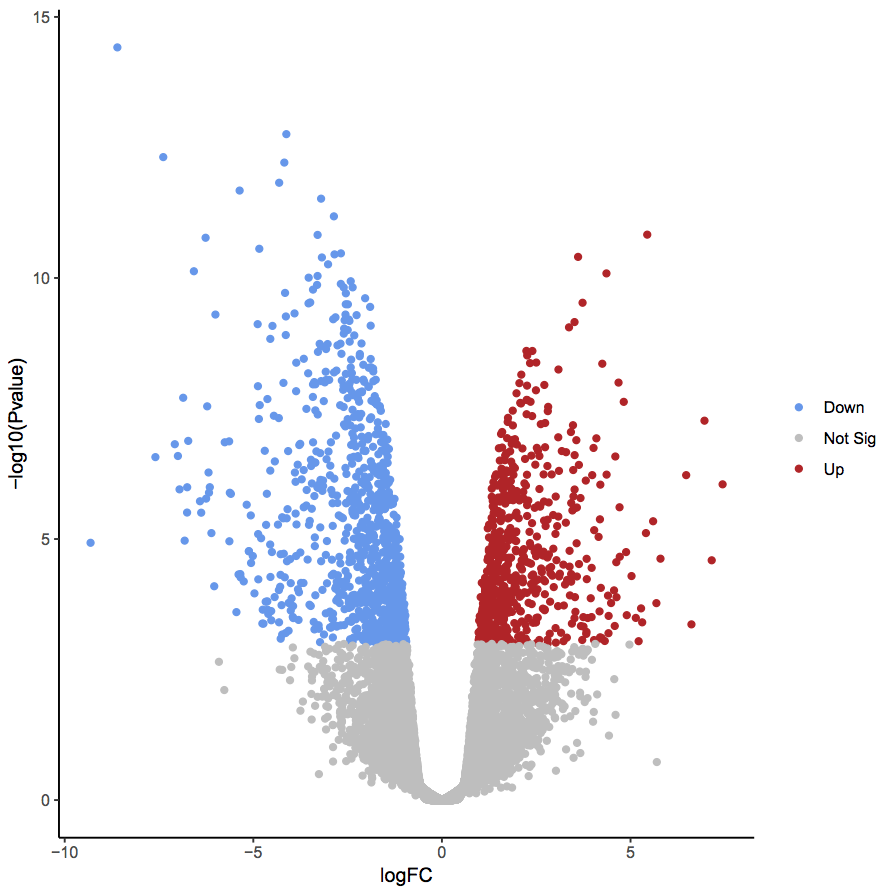

https://datavzrd.github.io
State of the art
Individual tables (tsv, excel) and plots:
- easy to publish
- limited interactivity
- no jumping between corresponding items
Web applications (custom, shiny, ...):
- running server (or local installation)
- implementation overhead
- long-term maintenance is challenging
https://datavzrd.github.io
The problem
Input:
- set of tables
- relations between tables
- set of rendering definitions
Output:
portable interactive visual presentation
https://datavzrd.github.io
datasets:
oscars:
path: "data/oscars.csv"
links:
link to oscar plot:
column: age
view: oscar-plot
link to movie:
column: movie
table-row: movies/Title
movies:
path: "data/movies.csv"
links:
link to oscar entry:
column: Title
table-row: oscars/movie
views:
oscars:
dataset: oscars
desc: |
### All winning oscars beginning in the year 1929.
This table contains *all* winning oscars for best
actress and best actor.
page-size: 25
render-table:
columns:
age:
plot:
ticks:
scale: linear
domain:
- 20
- 100
name:
link-to-url: "https://lmgtfy.app/?q=Is {name} in {movie}?"
movie:
link-to-url: "https://de.wikipedia.org/wiki/{value}"
award:
plot:
heatmap:
scale: ordinal
domain:
- Best actor
- Best actress
range:
- "#add8e6"
- "#ffb6c1"
index(0):
display-mode: hidden
regex('birth_.+'):
display-mode: detail
movies:
dataset: movies
render-table:
columns:
Genre:
ellipsis: 15
imdbID:
link-to-url: "https://www.imdb.com/title/{value}/"
Title:
link-to-url: "https://de.wikipedia.org/wiki/{value}"
imdbRating:
precision: 1
plot:
bars:
scale: linear
domain:
- 1
- 10
Rated:
plot-view-legend: true
plot:
heatmap:
scale: ordinal
color-scheme: accent
oscar-plot:
dataset: oscars
desc: |
## My beautiful oscar scatter plot
*So many great actors and actresses*
render-plot:
spec-path: ".examples/specs/oscars.vl.json"
movies-plot:
dataset: movies
desc: |
All movies with its *runtime* and *ratings* plotted over *time*.
render-plot:
spec-path: ".examples/specs/movies.vl.json"
The solution: Datavzrd
https://datavzrd.github.io
Felix Wiegand, P44
https://datavzrd.github.io
Portability
├── index.html
├── movies
│ ├── index_1.html
│ └── table.js
├── oscars
│ ├── index_1.html
│ └── table.js
├── movies-plot
│ └── index_1.html
├── oscar-plot
│ └── index_1.html
└── static
├── bootstrap.bundle.min.js
├── bootstrap.min.css
├── bootstrap-select.min.css
├── bootstrap-select.min.js
├── bootstrap-table-filter-control.min.js
├── bootstrap-table-fixed-columns.min.css
├── bootstrap-table-fixed-columns.min.js
├── bootstrap-table.min.css
├── bootstrap-table.min.js
├── datavzrd.css
├── jquery.min.js
├── jsonm.min.js
├── lz-string.min.js
├── showdown.min.js
├── vega-embed.min.js
├── vega-lite.min.js
└── vega.min.js- static HTML, no server needed
- load data via script tags
- javascript compliant compression
- \(\leq\) 30000 rows: in-memory filter/sort/paging
- \(>\) 30000 rows: paging/search projected to filesystem structure
https://datavzrd.github.io
Datavzrd + Snakemake
rule datavzrd:
input:
config="resources/{sample}.datavzrd.yaml",
table="data/{sample}.tsv",
output:
report(
directory("results/datavzrd-report/{sample}"),
htmlindex="index.html",
),
wrapper:
"v4.6.0/utils/datavzrd"https://datavzrd.github.io
Datavzrd + Snakemake
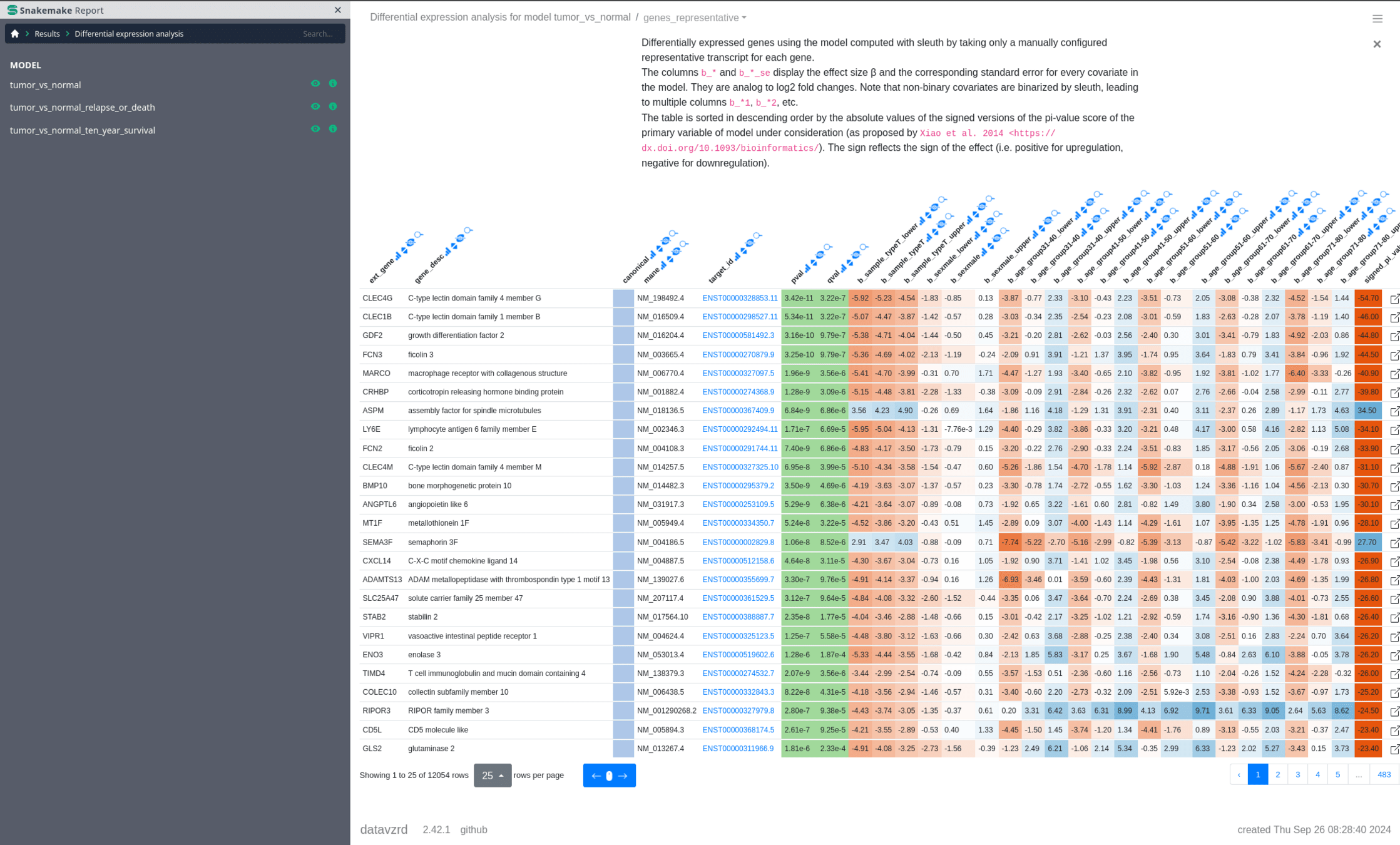
https://datavzrd.github.io
Varlociraptor
Varlociraptor
Johannes Köster, Louis Dijkstra, Tobias Marschall, Alexander Schönhuth.
Varlociraptor: enhancing sensitivity and controlling false discovery rate in somatic indel discovery. Genome Biology 21, 2020
https://varlociraptor.github.io
Traditional variant calling approach:
detection
genotyping/VAF prediction
Varlociraptor approach:
candidate variant calling
event calling
same tool
any tool
Varlociraptor

Varlociraptor
Johannes Köster, Louis Dijkstra, Tobias Marschall, Alexander Schönhuth.
Varlociraptor: enhancing sensitivity and controlling false discovery rate in somatic indel discovery. Genome Biology 21, 2020
https://varlociraptor.github.io
Varlociraptor
germline: "normal:0.5 | normal:1.0"
somatic_normal: "normal:]0.0,0.5["
somatic_tumor: "normal:0.0 & tumor:]0.0,1.0]"
absent: "normal:0.0 & tumor:0.0https://varlociraptor.github.io
Varlociraptor model building block
\(\xi_i \sim \text{Bernoulli}(\theta \tau)\)
\(\omega_i \sim Bernoulli(\pi_i)\)
\(Z_i \mid \xi_i, \omega_i=1,\beta,\delta \sim\)


\(\beta, \delta\)
- allele frequency
- sampling bias
- allele uncertainty
- biases/artifacts (strand, orientation, softclip, homopolymer, ...)
- alignment uncertainty

https://varlociraptor.github.io
Variant calling grammar
species:
heterozygosity: 0.001
ploidy:
male:
all: 2
X: 1
Y: 1
female:
all: 2
X: 2
Y: 0
samples:
jane:
sex: femaleevents:
present: "jane:0.5 | jane:1.0"https://varlociraptor.github.io
species:
heterozygosity: 0.001
ploidy:
male:
all: 2
X: 1
Y: 1
female:
all: 2
X: 2
Y: 0
samples:
jane:
sex: female
john:
sex: male
james:
sex: male
inheritance:
mendelian:
mother: jane
father: johnevents:
john: "john:0.5 | john:1.0"
jane: "jane:0.5 | jane:1.0"
denovo_james: "(james:0.5 | james:1.0) & !$jane & !$john"Variant calling grammar
https://varlociraptor.github.io
species:
heterozygosity: 0.001
ploidy:
male:
all: 2
X: 1
Y: 1
female:
all: 2
X: 2
Y: 0
samples:
jane:
sex: female
somatic-effective-mutation-rate: 1e-10
tumor:
inheritance:
clonal:
from: jane
contamination:
by: jane
fraction: 0.1
somatic-effective-mutation-rate: 1e-6events:
germline: "jane:0.5 | jane:1.0"
somatic: "jane:]0.0,0.5["
somatic_tumor_low: "jane:0.0 & tumor:]0.0,0.1["
somatic_tumor_high: "jane:0.0 & tumor:[0.1,1.0]"
Variant calling grammar
https://varlociraptor.github.io
samples:
jane:
sex: female
somatic-effective-mutation-rate: 1e-10
tumor:
inheritance:
clonal:
from: jane
contamination:
by: jane
fraction: 0.1
somatic-effective-mutation-rate: 1e-6
relapse:
inheritance:
clonal:
from: jane
contamination:
by: jane
fraction: 0.2
somatic-effective-mutation-rate: 1e-6expressions:
somatic_tumor: "jane:0.0 & tumor:]0.0,1.0]"
events:
germline: "jane:0.5 | jane:1.0"
somatic: "jane:]0.0,0.5["
somatic_tumor_no_increase: "$somatic_tumor & l2fc(relapse,tumor) < 1"
somatic_tumor_increase: "$somatic_tumor & l2fc(relapse,tumor) >= 1"
somatic_relapse: "jane:0.0 & tumor:0.0 & relapse:]0.0,1.0]"
Variant calling grammar
https://varlociraptor.github.io
species:
heterozygosity: 0.001
ploidy:
male:
all: 2
X: 1
Y: 1
female:
all: 2
X: 2
Y: 0
samples:
dna_illumina:
sex: female
dna_nanopore:
inheritance:
clonal:
from: dna_illumina
rna_illumina:
universe: [0.0,1.0]events:
het: "dna_illumina:0.5 & dna_nanopore:0.5 & rna_illumina:]0.0,1.0]"
hom: "dna_illumina:1.0 & dna_nanopore:1.0 & rna_illumina:1.0"
rna_editing: "dna_illumina:0.0 & dna_nanopore:0.0 & rna_illumina:]0.0,1.0]"
Variant calling grammar
https://varlociraptor.github.io
Long read support
https://varlociraptor.github.io


Discovering nucleotide level and structural variants in cancer genome data from second and third generation sequencing technologies. Till Hartmann, PhD thesis 2024
Continuous testing
varlociraptor call variants --testcase-prefix testcase --testcase-locus CHROM:POS generic \
--scenario scenario.yaml --obs tumor=tumor.bcf normal=normal.bcfAutomatic test case generation:
145
public testcases (simulated + real benchmarks)
66
private testcases
(clinical)
https://varlociraptor.github.io

Reporting with Datavzrd
https://varlociraptor.github.io
Beyond just variant calling
Haplotype quantification:
utilizing Varlociraptor's using posterior allele frequency distributions [Hamdiye Uzuner, talk today]
Methylation calling:
joint consideration of methylation and variation, any sequencing technology [Adrian Prinz, P40]
Optical mapping evidence:
extension of statistical model to consider optical mapping label positions to determine SV allele likelihoods [Laura Kühle]
Phased variant impact prediction:
leveraging matching between Varlociraptor observations and variants [Felix Wiegand]
Fusion calling:
using generic breakends [Felix Mölder]
Acknowledgements
Koesterlab
Laura Kühle
David Lähnemann
Felix Mölder
Adrian Prinz
Hamdiye Uzuner
Felix Wiegand
Can Özkan
Andrea Tonk
Till Hartmann (alumn.)
Snakemake Community
Christian Meesters
Michael B. Hall
Filipe G. Vieira
Morten E. Lund
Vanessa Sochat
Alexaner Kanitz
Kim-Phillip Jablonski
Brice Letcher
Michael B. Hall
Chris Tomkins-Tinch
Sven O. Twardziok
Manuel Holtgrewe
+ hundreds of contributors
Bioconda Community
Björn Grüning
Ryan Dale
Marcell Bargull
Brad Chapman
Chris Tomkins-Tinch
Andreas Sjödin
Devon Ryan
Elmar Pruesse
Robert A. Petit III
Christian Brueffer
Alicia Evans
+ hundreds of contributors
Others
Sven Rahmann
Alex Schönhuth
Shirley Liu
Myles Brown
Henry Long
Louis Dijkstra
Tobias Marschall
Marcel Martin
Sven Nahnsen
Alex Schramm
Ina Pretzell
Michael Wessolly
Thomas Herold
Martin Schuler
all users of Snakemake, Bioconda, Datavzrd, and Varlociraptor

Snakemake hackathon 2025
March 10-14 at
Work on:
- plugins
- wrappers
- workflows
- internals

Visit LHC:
And shape the future of the ecosystem!
Registration will open soon, email me to get a notification: johannes.koester@uni-due.de
free lodging sponsored by FAIROS-HEP!
Keynote GCB 2024
By Johannes Köster




