


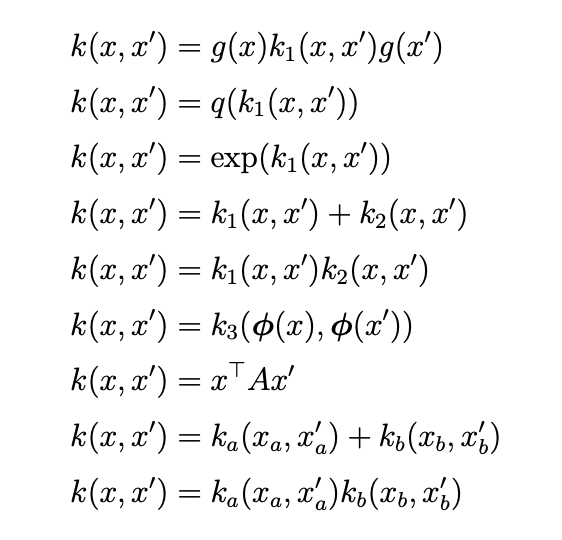
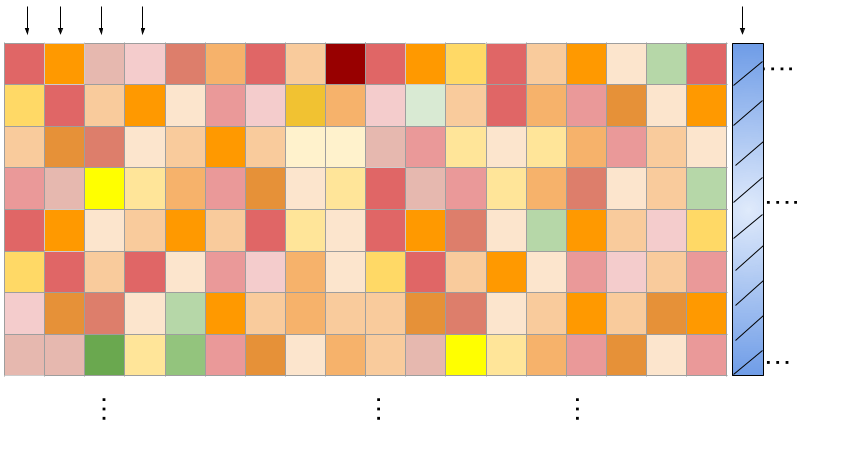
Vidhi Lalchand
Postdoctoral Fellow, Broad and MIT
Vidhi Lalchand
14-11-2025
Drug-synergy modelling through dose-response surfaces
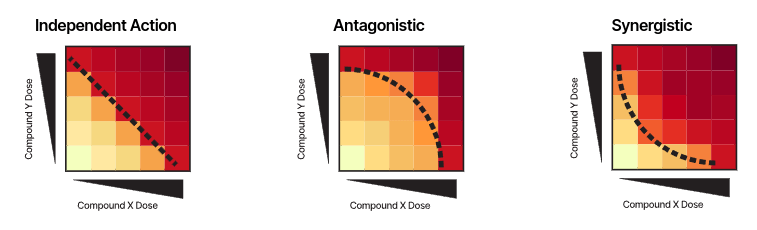
The nature of a drug combination, as synergistic, antagonistic or non-interacting, is usually studied by in-vitro dose–response experiments on cancer cell lines, through e.g. a cell viability assay.
https://theprismlab.org/white-papers/multiplexed-cancer-cell-line-combination-screening-using-prism
Drug-synergy modelling: Bliss model of independence
https://theprismlab.org/white-papers/multiplexed-cancer-cell-line-combination-screening-using-prism
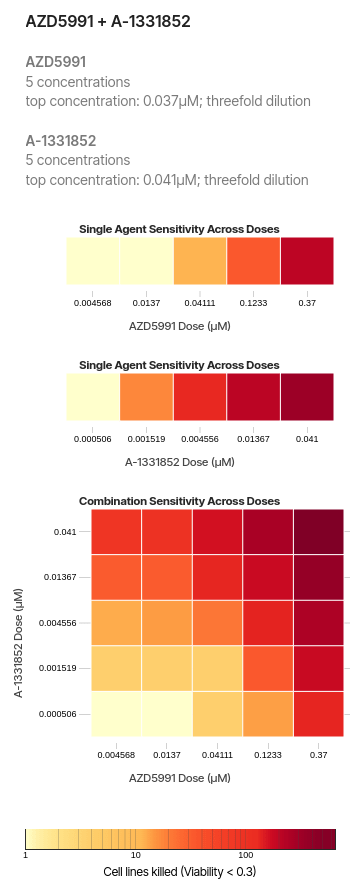
The Bliss model assumes that the two drugs AAX and Y act independently to achieve their effects.
EAE_AEX: The probability of drug AAA killing a cell.
EBE_BEY: The probability of drug BBB killing a cell.
The probability of neither drug killing a cell is:
(1−EA)⋅(1−EB).(1 - E_A) \cdot (1 - E_B).(1−EX)⋅(1−EY)Therefore, the expected viability \(V_{XY}\) is,VABV_{ABVunder independence is:
\(V_{X} = 0.5\) and \(V_{Y} =0.6\)
\(V_{XY} = 0.3\)
Drug-synergy modelling through dose-response surfaces
Bliss independence
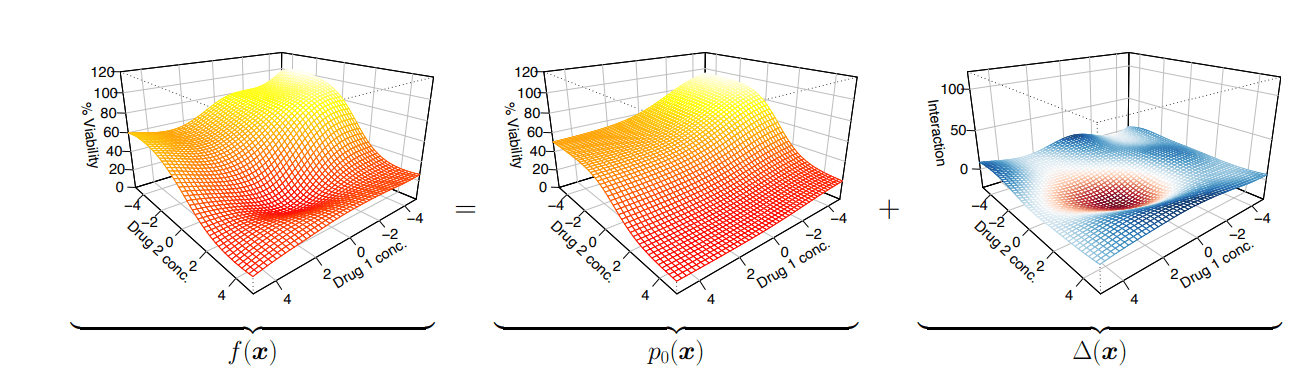
Non-linear effect
I use Gaussian processes to model this non-linear effect
What is a GP?
A sample from a \(k\)-dimensional Gaussian \( \mathbf{x} \sim \mathcal{N}(\mu, \Sigma) \) is a vector of size \(k\). $$ \mathbf{x} = [x_{1}, \ldots, x_{k}] $$
The mathematical crux of a GP is that \( \textbf{f} \equiv [f(x_{1}), f(x_{2}), f(x_{3}),....., f(x_{N})]\) is just a N-dimensional multivariate Gaussian.
A GP is an infinite dimensional analogue of a Gaussian distribution \( \rightarrow \) a sample from it is a vector of infinite length?
But at any given point, we only need to represent our function \( f(x) \) at a finite index set \( \mathcal{I} = [x_{1},\ldots, x_{500}]\). So we are interested in our 500 dimensional function vector \( [f(x_{1}), f(x_{2}), f(x_{3}),....., f(x_{500})]\).
Marginalising function values corresponding to inputs not in our index set.
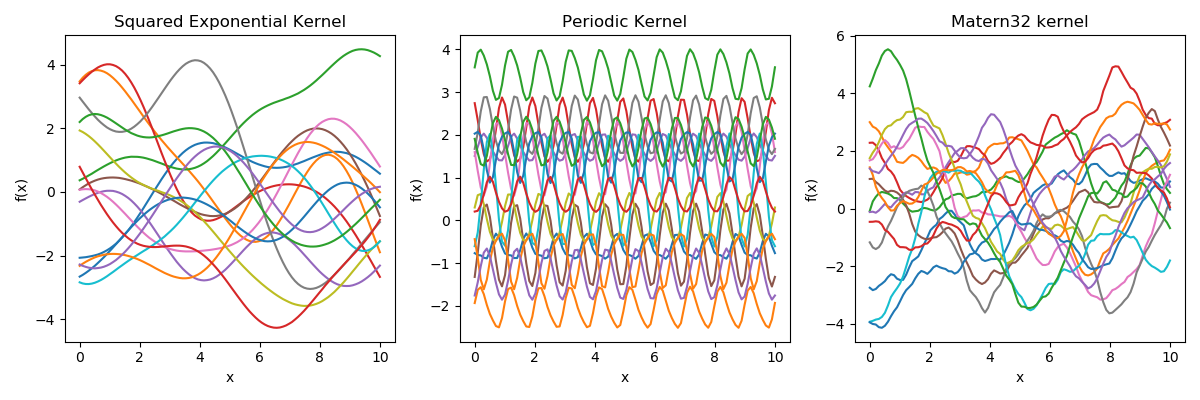
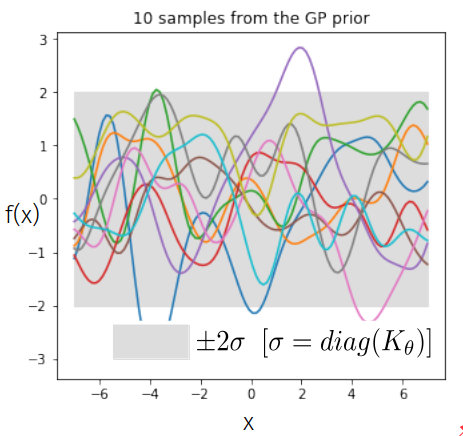
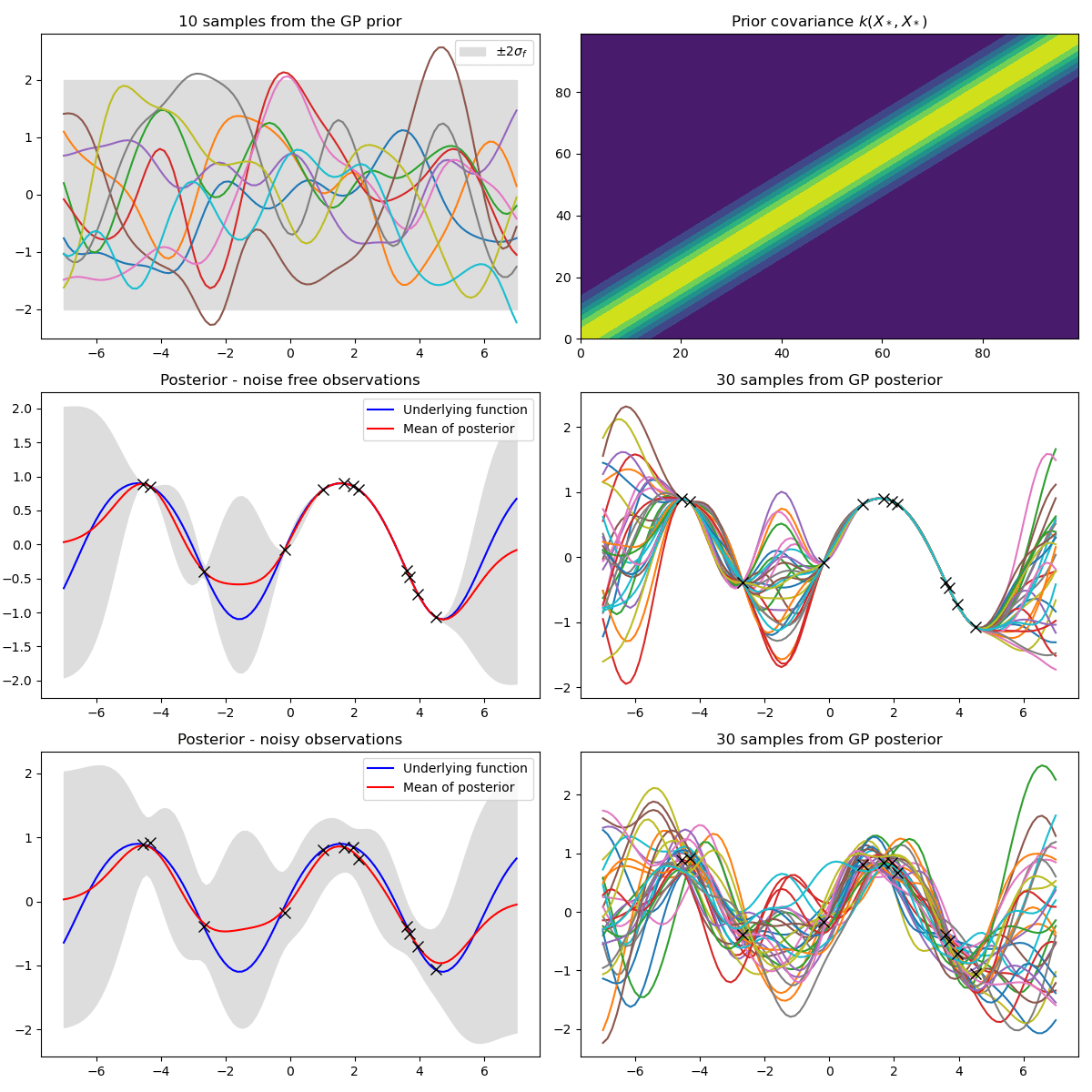
Samples from a GP
prior over functions \( \rightarrow \)
Sample draws from a zero mean GP prior under different kernel functions.
In reality, they are just draws from a multivariate Gaussian \( \mathcal{N}(0, K)\) where the covariance matrix has been evaluated by applying the kernel function to all pairs of data points.
Rigorous definition: \( f\) is a GP if for any finite subset \(\{x_{1}, \ldots, x_{n}\} \subset \mathcal{X}\), the marginal distribution of function values \( p(\textbf{f})\) is Gaussian.
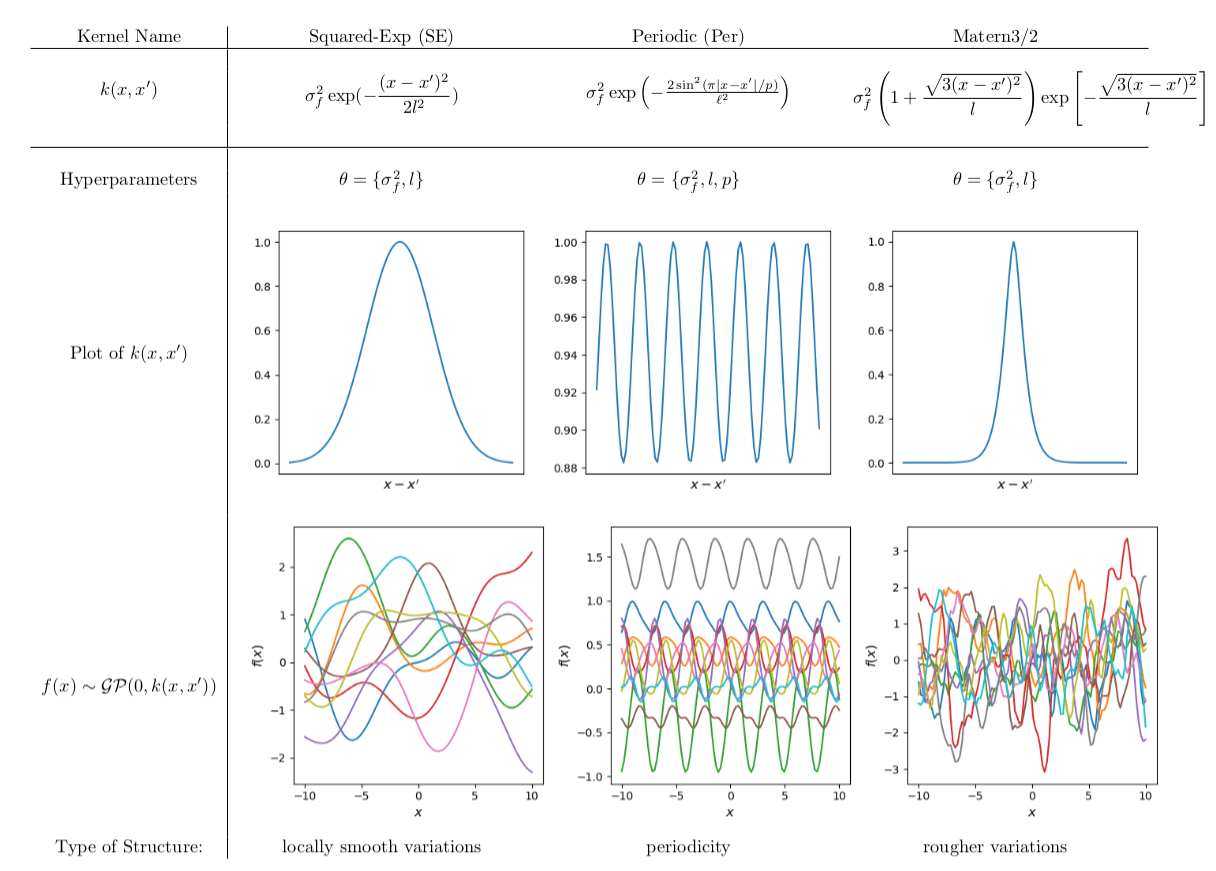
Popular Kernels
Usually, the user picks one on the basis of prior knowledge.
Each kernel depends on some hyperparameters \( \theta\), which are tuned in the training step.
The kernel function \( k(x,x')\) is the heart of a GP, it controls all of the inductive biases in our function space like shape, periodicity, smoothness.
Closed operations on kernels
Stationary kernels
Gaussian Process Regression
How do we fit functions to noisy data with GPs?
1. Given some noisy data \( \bm{y} = \lbrace{y_{i}}\rbrace_{i=1}^{N} \) at \( X = \{ x_{i}\}_{i=1}^N\) input locations.
2. You believe your data comes from a function \( \bm{f}\) corrupted by Gaussian noise.
$$ y_{i} = f(x_{i}) + \epsilon, \hspace{10pt} \epsilon \sim \mathcal{N}(0, \sigma^{2})$$
Data Likelihood: \( \hspace{10pt} \bm{y}|\bm{f} \sim \mathcal{N}(\bm{f}, \sigma^{2}\mathbb{I}) \)
Prior over functions: \( f|\theta \sim \mathcal{GP}(0, k_{\theta}), \bm{f} \sim \mathcal{N}(\bm{0}, K_{\theta}) \)
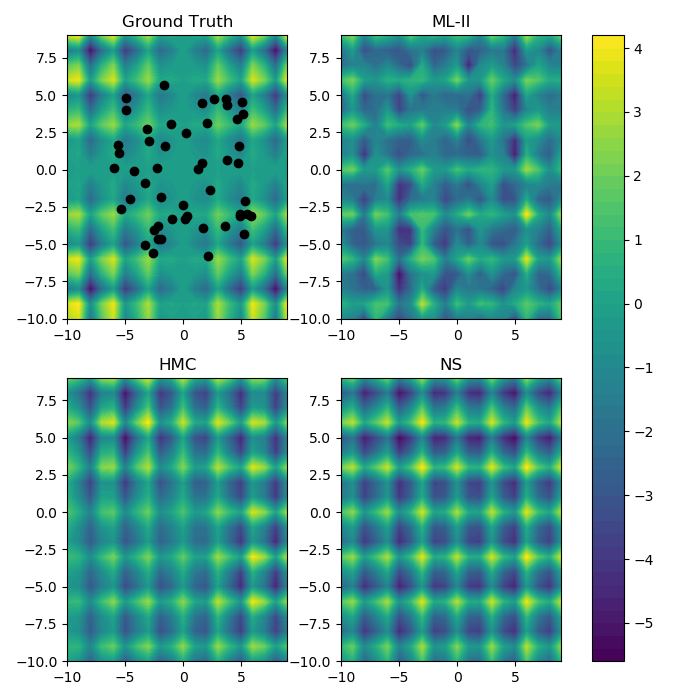
Example of fitting a 2d surface with GP regression
Multiple inference methods
Ground Truth
GPs are excellent for fitting low dimensional surfaces and adapting to the smoothness characteristics inherent in the data.
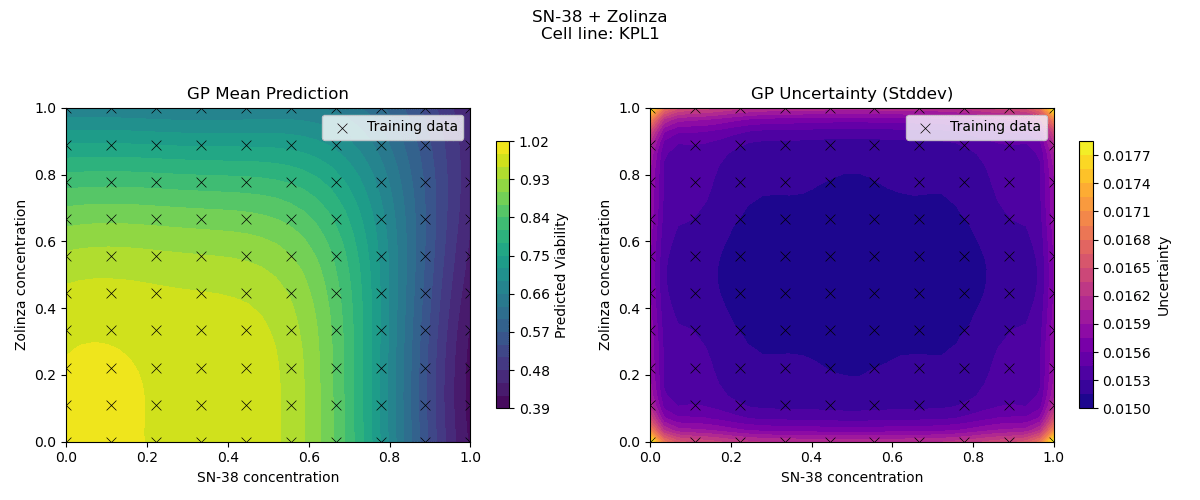
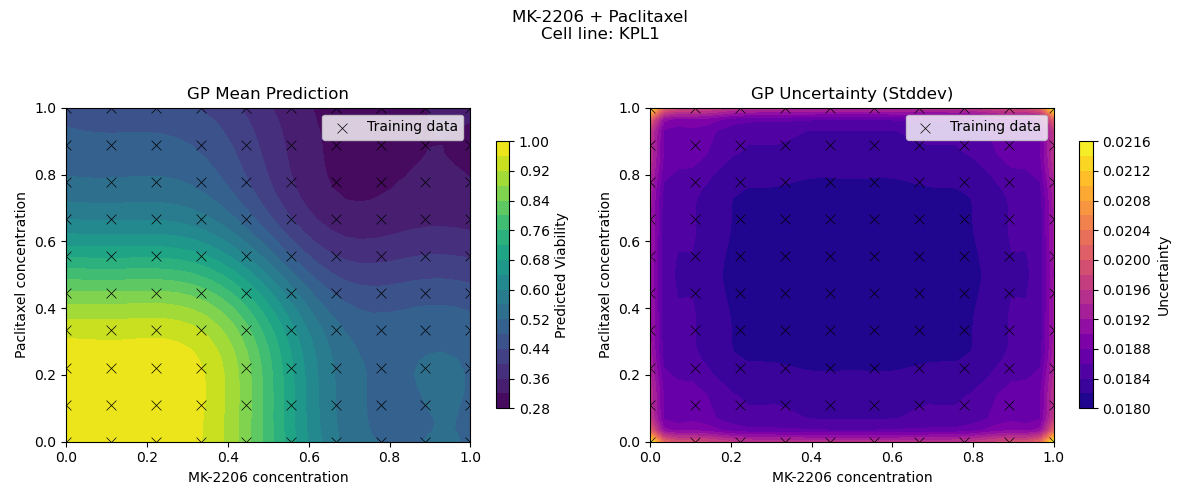
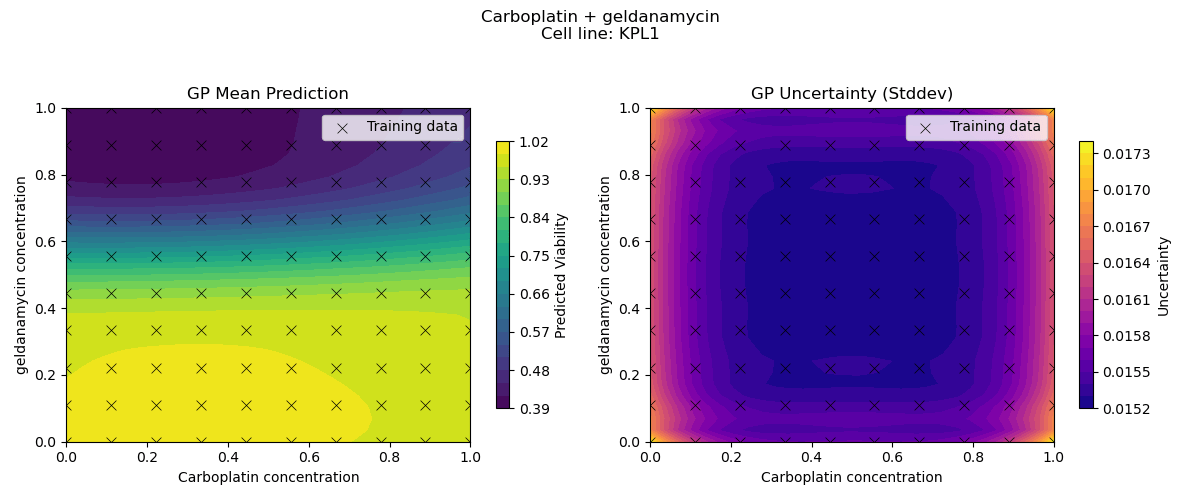
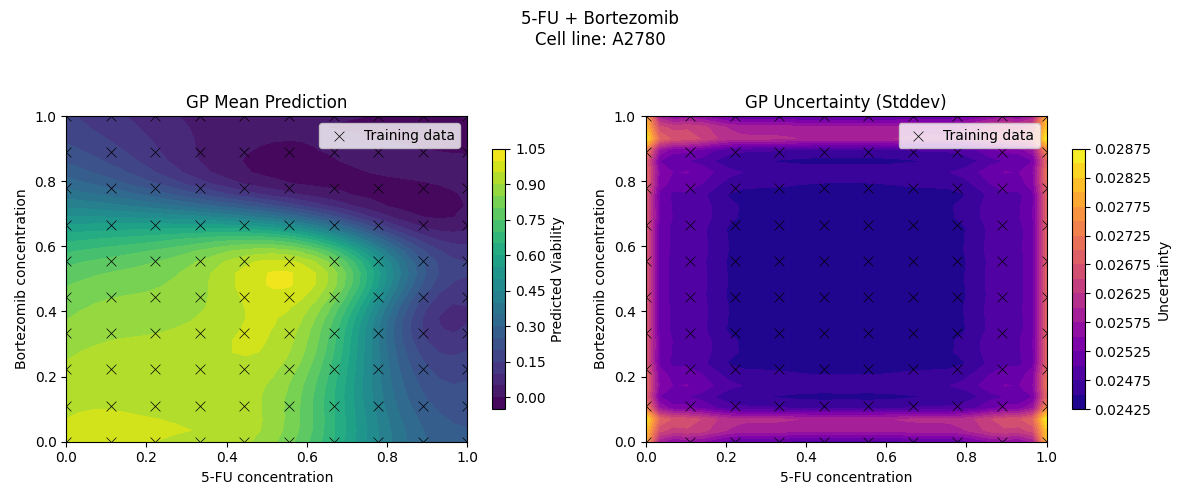
Sample drug synergy surfaces [2d fitted with a GP]
Computational Problem
Learn a model from observed drug combination viabilities
Predict a drug synergy surface for unseen chemical compounds
[For simplicity, we fix the cell-line and first try to model the variation in viabilities driven by variation in drugs]
(c1, A, B)
(c1, C, D)
(c1, E, F)
(c1, X, Y)
(0, 0.11)
(0,0.22)
(0,0.33)
........
(1,1)
\(N_{d}\) = 583
\(N_{}\) = 100
No. of drug concentrations
No. of drug pairs.
Mathematical Framework (1/2)
Step 2: Find an optimal basis
Step 1:
is the matrix whose columns are the shared basis functions,
is the matrix of coefficients for each drug pair, column-wise
The minimisation has a closed-form solution in the form of SVD:
This guarantees the smallest reconstruction error among all orthonormal bases of size KKK
Mathematical Framework (2/2)
Rows of YYY : sampled points on the surface (shared grid).
Columns of YYY : drug pairs.
Columns of Φ\PhiΦ : basis functions (shared spatial patterns).
Rows of CCC : coefficients describing how much each pattern appears in each pair.
Step 3: Regress on to the coefficients from the drug embeddings:
Step 4: Reconstruct surface from the predicted coefficients for each pair
For a new drug pair p\*p^\*p*:
You compute its drug embedding ψ(A\*,B\*)\psi(A^\*,B^\*)ψ(A*,B*)
Predict the coefficient vector \( \hat{c}_{p_{*}}\) via your regression model hhh.
Reconstruct the full surface,
Note: Choice of drug embeddings should be permutation invariant
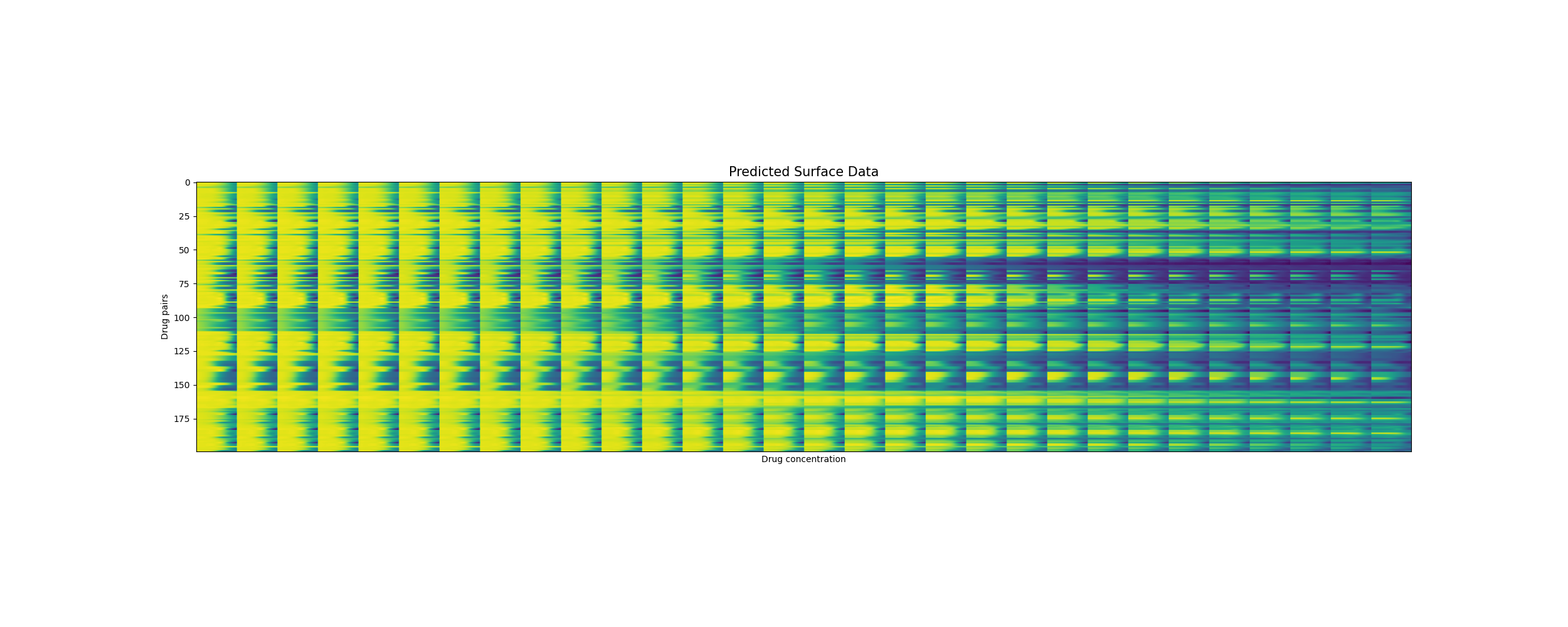
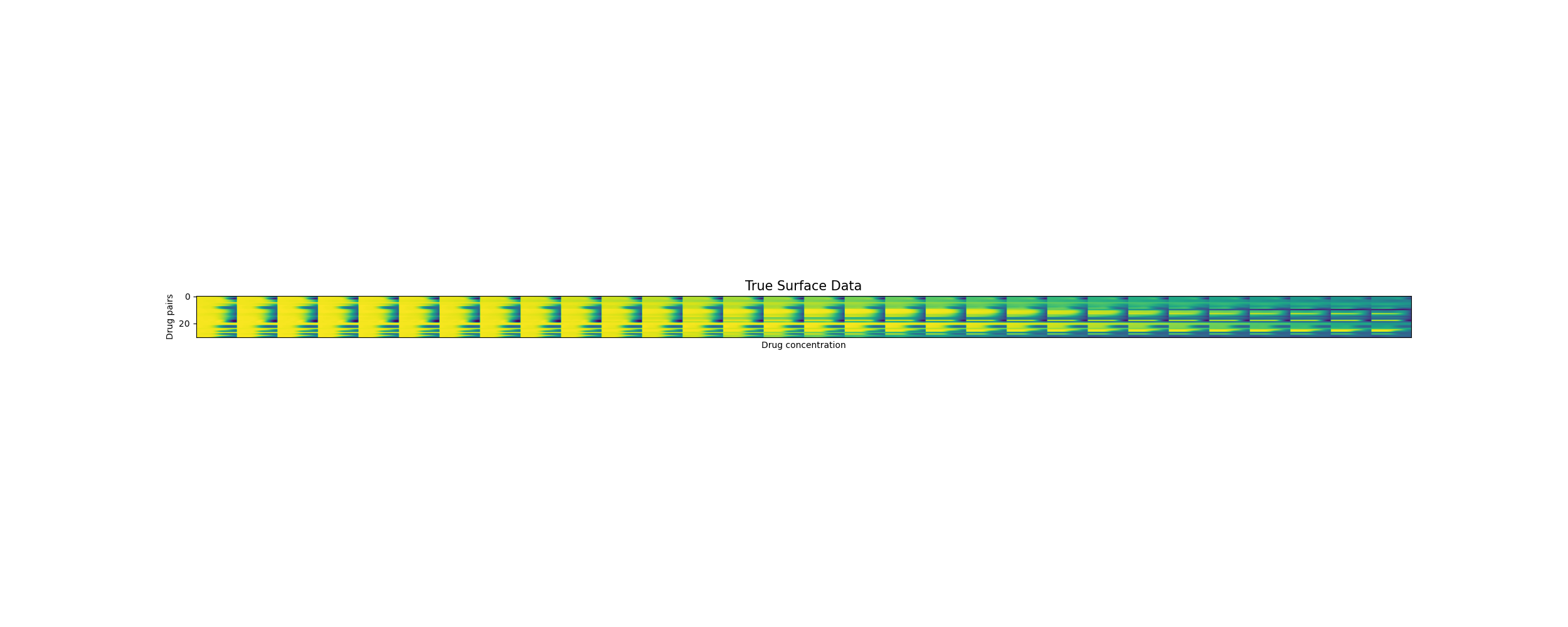
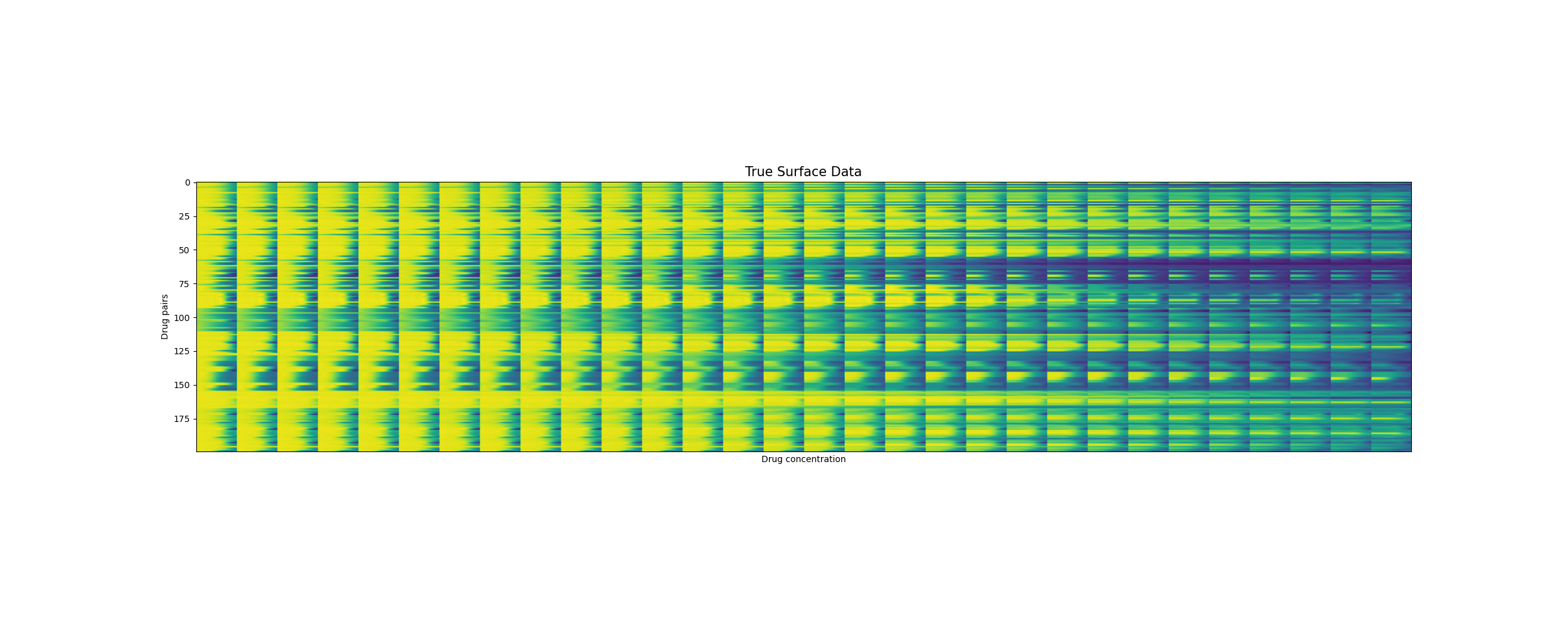
Predicting unseen drug surfaces (Y vs. \(\hat{Y}\))
Drug pairs
Drug concentrations
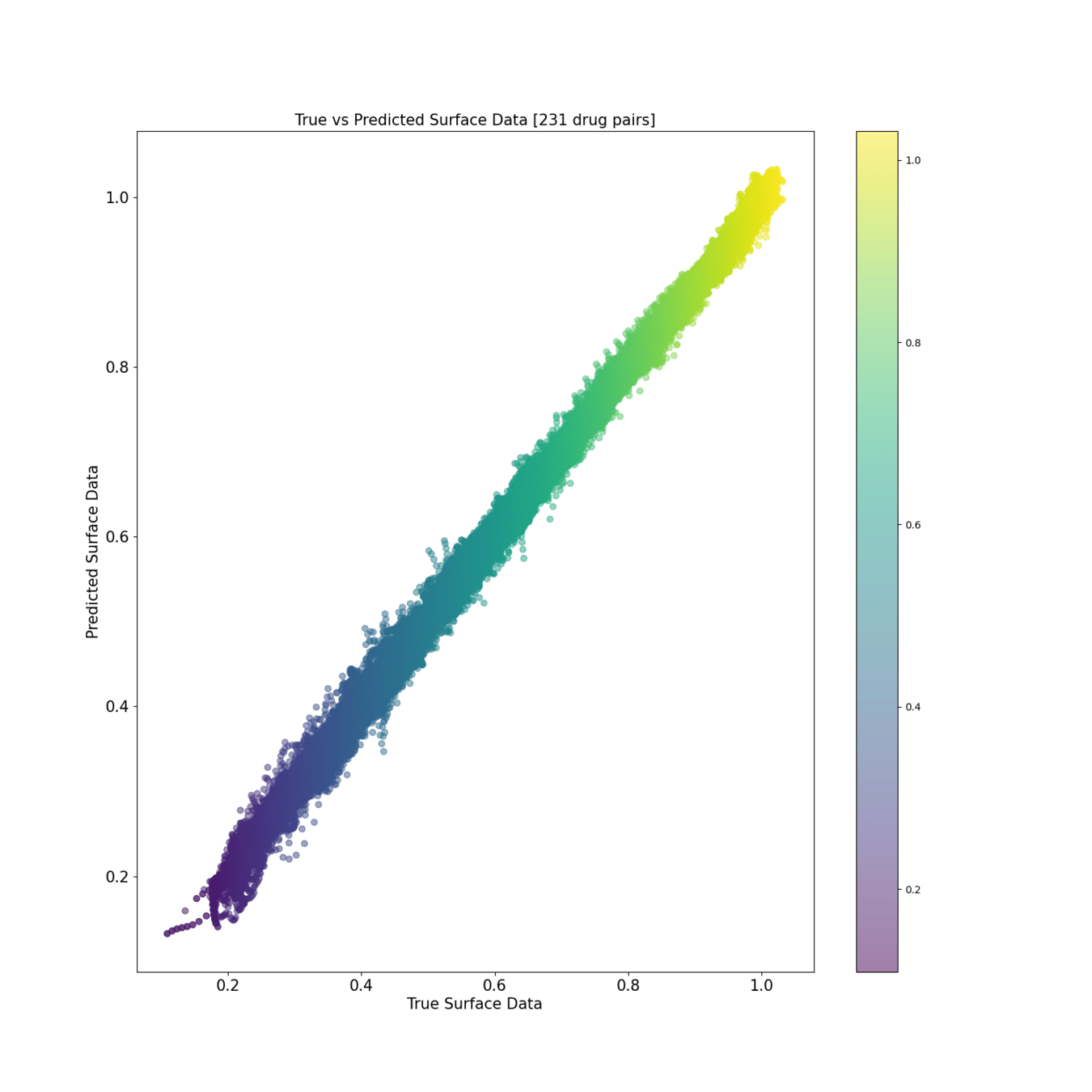
Training reconstruction [200 drug pairs]
Flattened drug surfaces
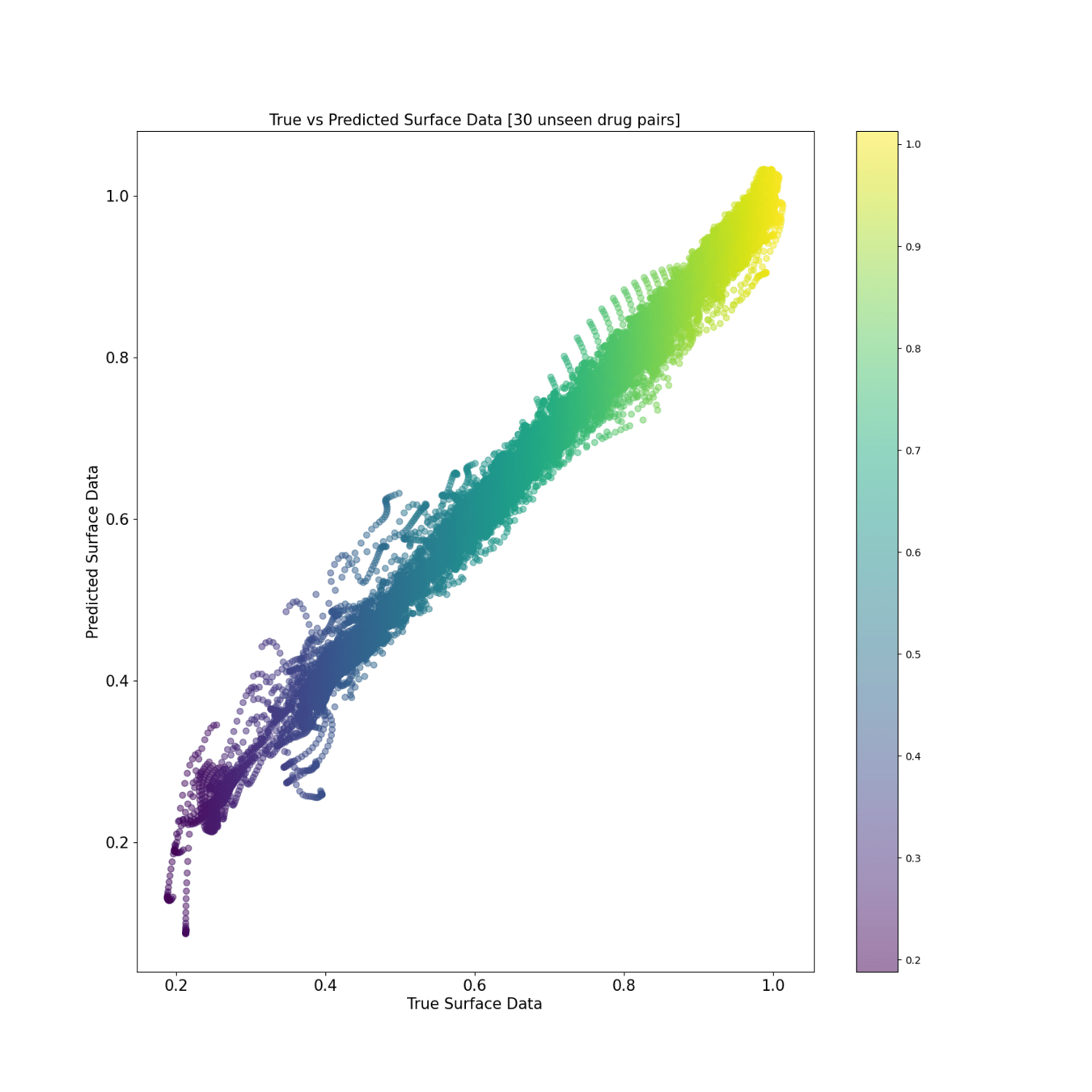
Test reconstruction [30 unseen pairs]
Scatter view of predicted drug synergies: Training and Test
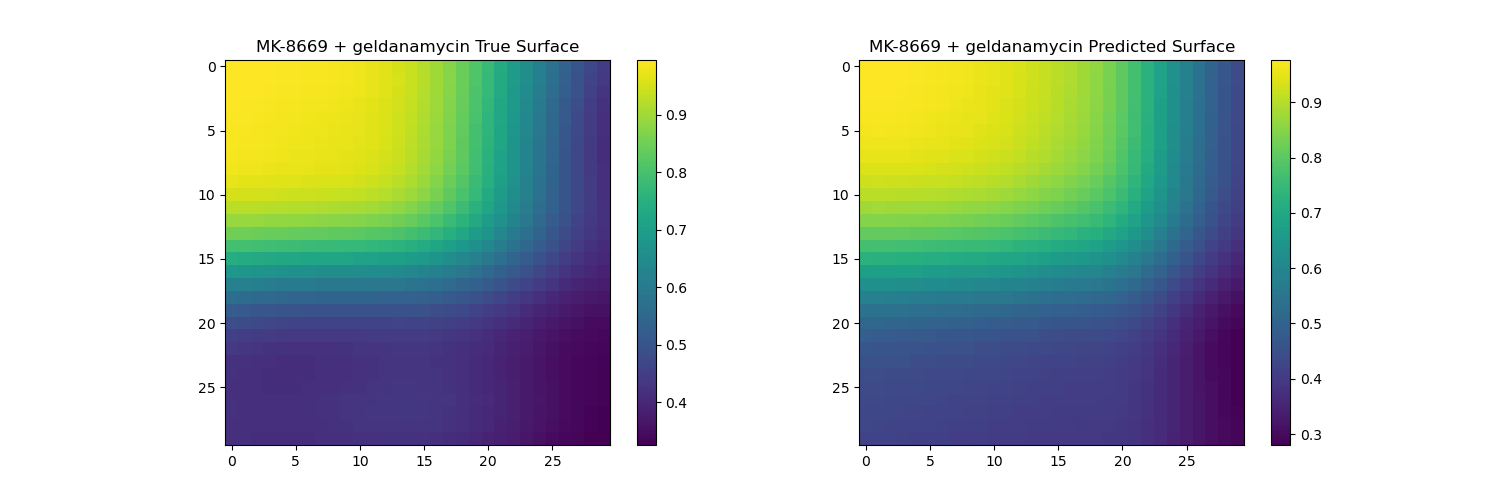
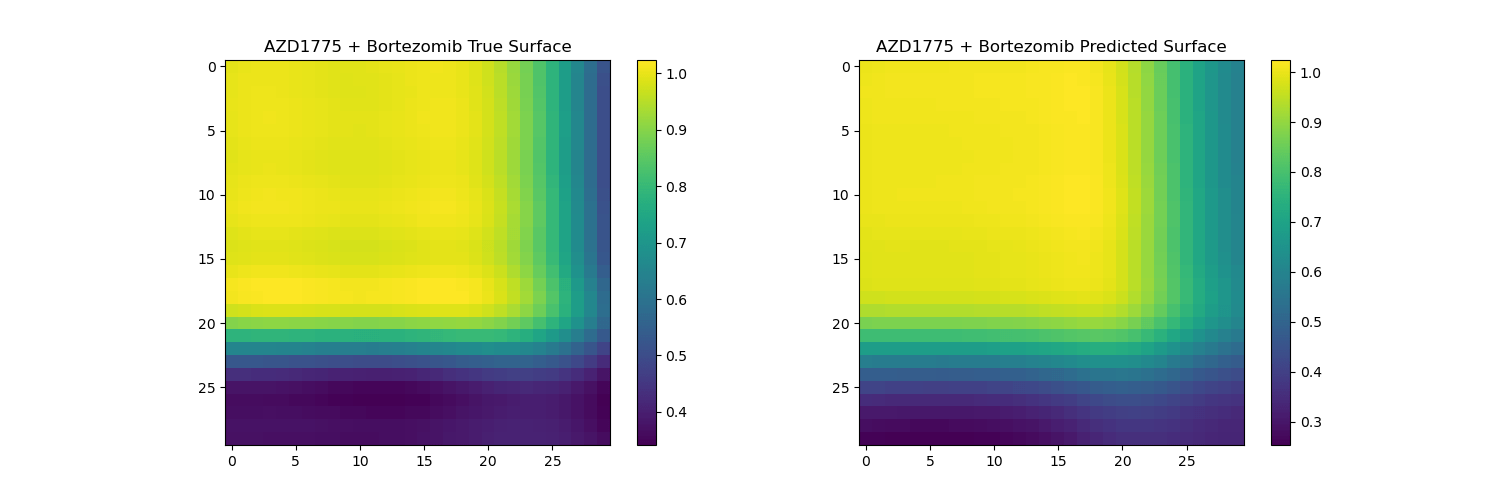
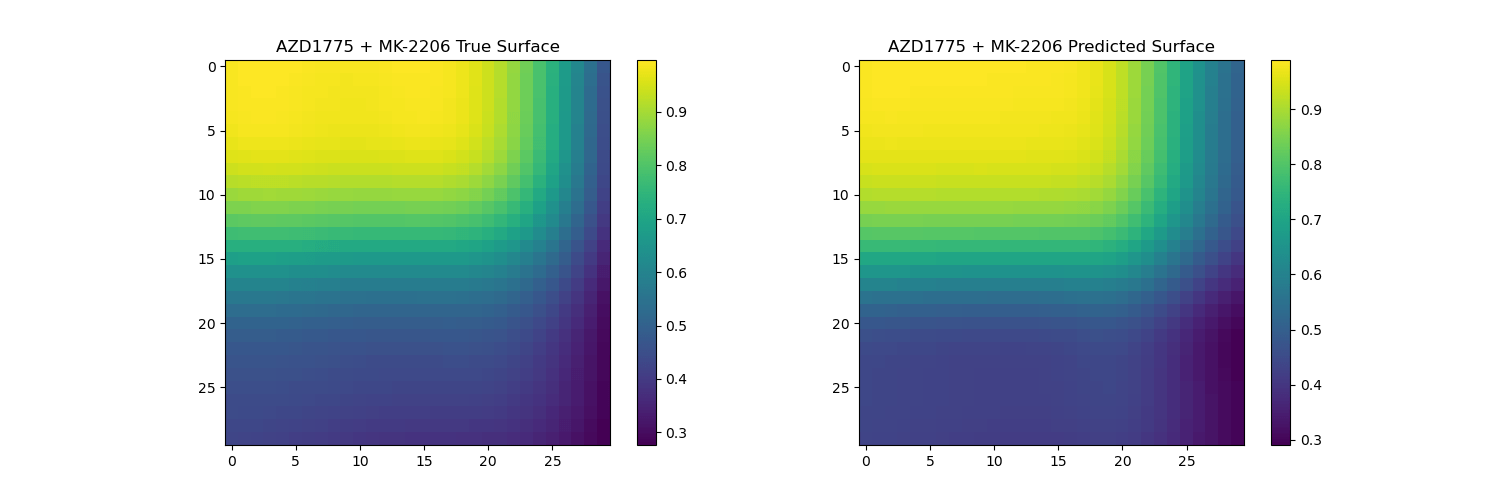
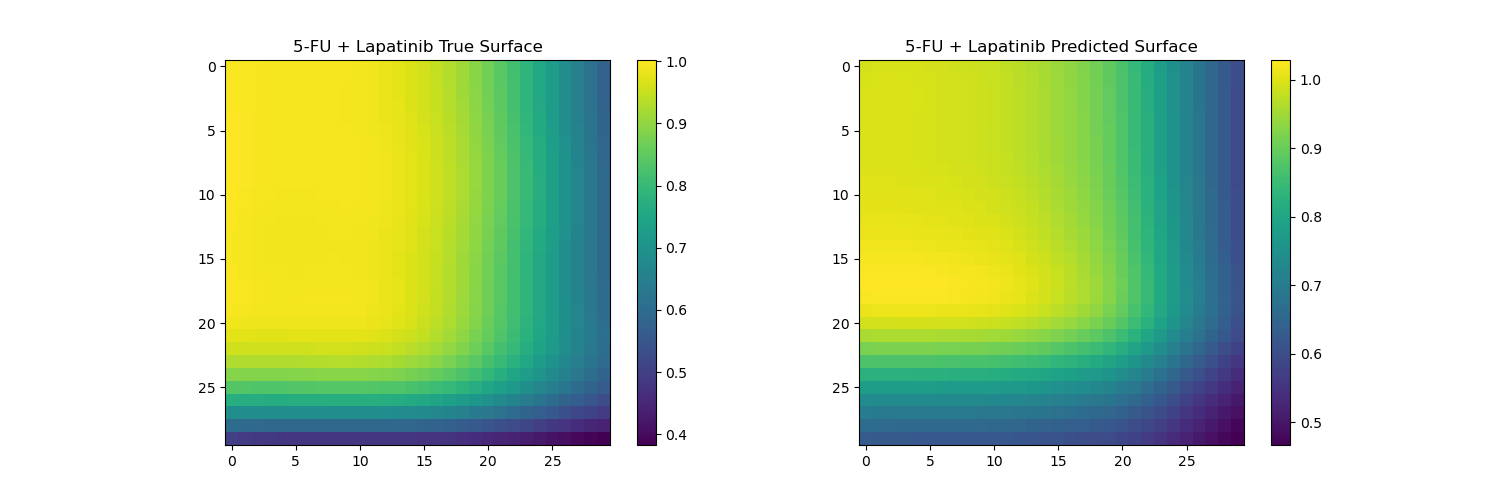
Surface reconstruction from coefficients on unseen drugs
Surface reconstruction from coefficients on unseen drugs
The predicted surfaces are good at capturing the overall shape of the surface but their variations are smoother than the real viabilities.
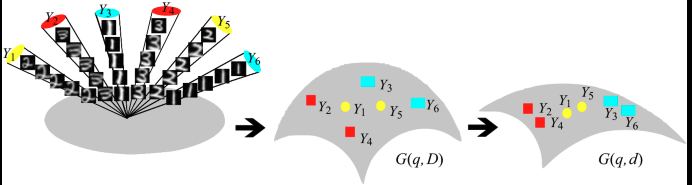
Extensions: Geometry view
Thank you!
What about generalising to unseen cell-lines (harder than for drugs, more variation)?
When you run SVD, you are selecting a point on the Grassmann manifold,
Gr(K, D),\mathrm{Gr}(K,\,D),Gr(K,D),
the set of all KKK-dimensional linear subspaces of a DDD-dimensional ambient space (D = G x G).
So the PCA subspace is a single point \([U*]\) on Gr(K,D)[U⋆][U_\star on Gr(K,D)\mathrm{Gr}(Gr(K,D)
The space Gr(K,D)\mathrm{Gr}(K,D)Gr(K,D) is not Euclidean; its elements are subspaces but what is useful about this view is that it gives a natural metric for comparing subspaces, distances between subspaces, interpolation and traversal.
If later you learn a new subspace UnewU_\text{new}Unew (say, from new data or another cell line), you can treat both as points on Gr(K,D)\mathrm{Gr}(K,D)Gr(K,D) and study their alignment via principal angles, this tells you whether the shape manifold of responses has shifted.
By Vidhi Lalchand



