


























Inês Mendes
Bioinformatics PhD student.
Inês Mendes
MRamirez Lab - iMM João Lobo Antunes
Instituto Gulbenkian de Ciência
14 July 2022
World Health Organisation Global Priority Pathogens list. This catalogue includes, besides Mycobacterium tuberculosis considered the number one global priority, a list of twelve microorganisms grouped under three priority tiers according to their antimicrobial resistance: critical (Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacteriaceae), high (Enterococcus faecium, Helicobacter pylori, Salmonella species, Staphylococcus aureus, Campylobacter species and Neisseria gonorrhoeae), and medium (Streptococcus pneumoniae, Haemophilus influenzae and Shigella species). The major objective was to encourage the prioritisation of funding and incentives, align research and development priorities of public health relevance, and garner global coordination in the fight against antimicrobial-resistant bacteria. Adapted from World Health Organization, 2017.
Bacterial Population Genetics
Pathogenesis and Natural History of Infection
Outbreak Investigation and Control
Surveillance of Infectious Diseases
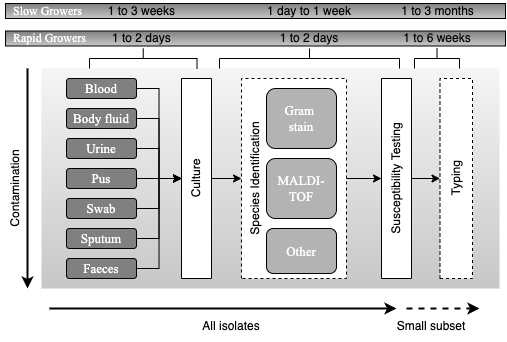
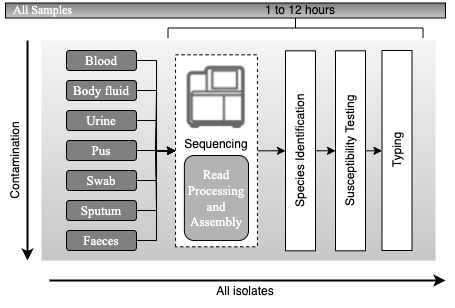
Principles of current processing of bacterial pathogens. Schematic representation of the current workflow for processing samples for bacterial pathogens is presented, with high complexity and a typical timescale of a few weeks to a few months. Samples that are likely to be normally sterile are often cultured on rich medium that will support the growth of any culturable organism. Samples contaminated with colonising flora present a challenge for growing the infecting pathogen. Many types of culture media (referred to as selective media) are used to favour the growth of the suspected pathogen. Once an organism is growing, the likely pathogens are then processed through a complex pathway that has many contingencies to determine species and antimicrobial susceptibility. Broadly, there are two approaches. One approach uses MALDI-TOF for species identification prior to setting up susceptibility testing. The other uses Gram staining followed by biochemical testing to determine species; susceptibility testing is often set up simultaneously with doing biochemical tests. Lastly, depending on the species and perceived likelihood of an outbreak, a small subset of isolates may be chosen for further investigation using a wide range of typing tests. Adapted from Didelot et al., 2012
Joana Silva, MRamirez Lab
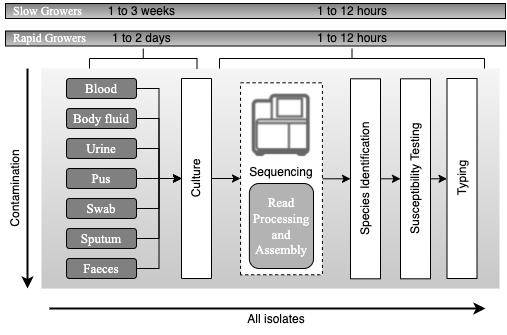
Principles of current processing of bacterial pathogens based on whole genome sequencing. Schematic representation of the workflow for processing samples for bacterial pathogens after the adoption of whole genome sequencing, with an expected timescale that could fit within a single day. The culture steps would be the same as currently used in a routine microbiology laboratory (see Figure 1.2). Once a likely pathogen is ready for sequencing, DNA is extracted, taking as little as 2 hours to prepare the DNA for sequenc- ing. After sequencing, the main processes for yielding information is computational. Automated sequence assembly algorithms are necessary for processing the raw sequence data, from which species, relationship to other isolates of the same species, antimicrobial resistance profile and virulence gene content can be assessed. All the results can also be used for outbreak detection and infectious diseases surveillance. Adapted from Didelot et al., 2012
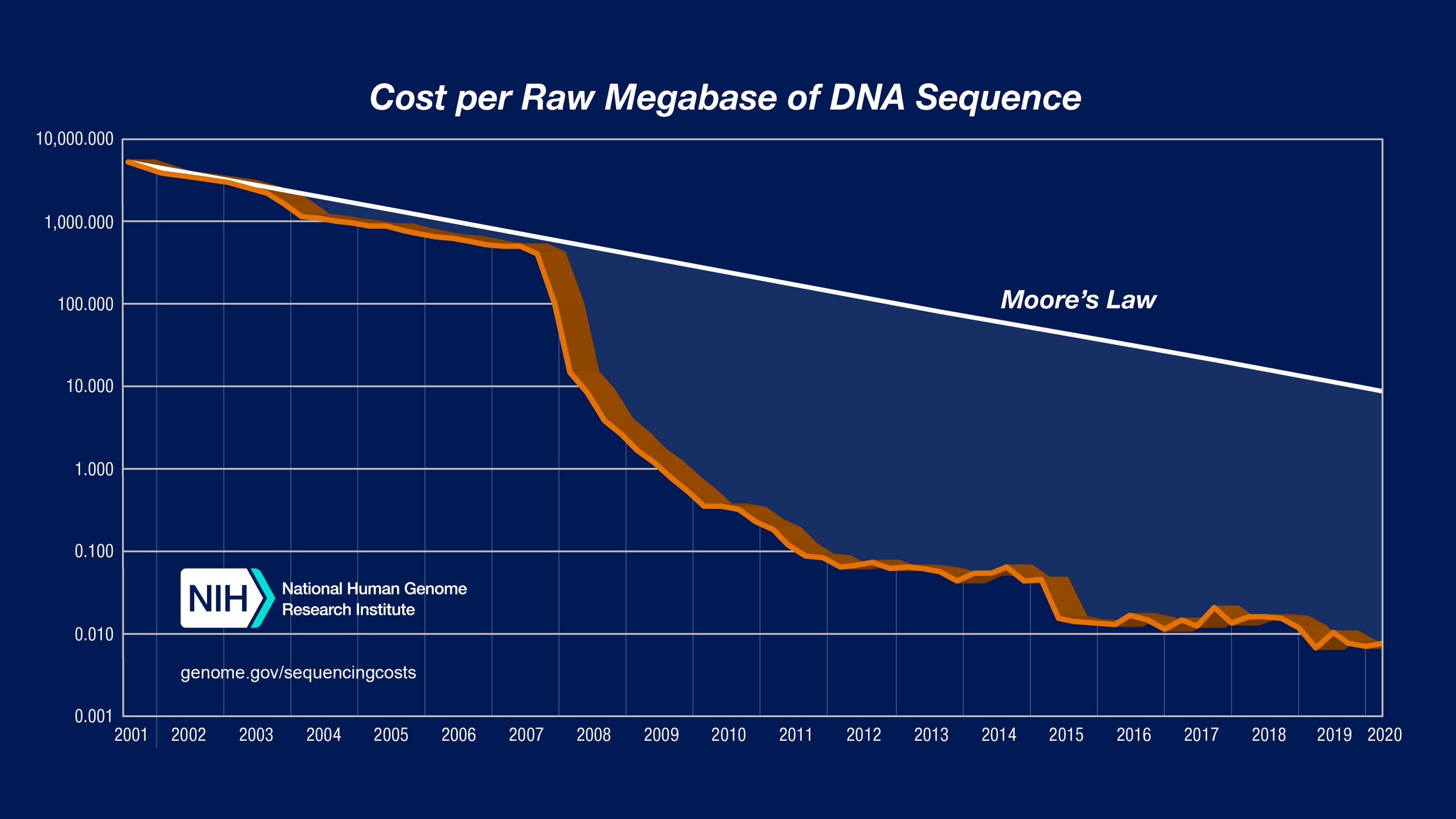
https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data
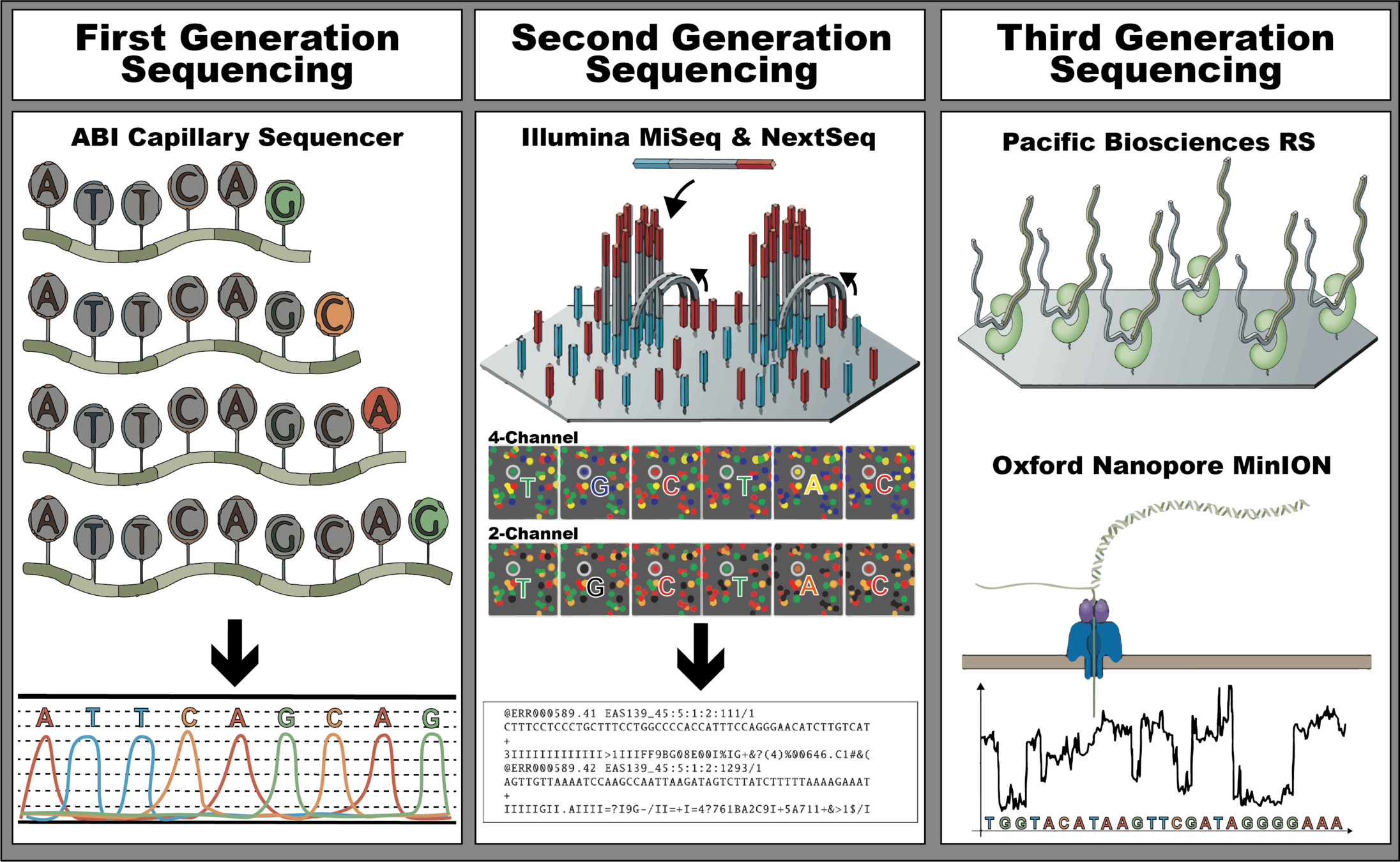
The three revolutions in sequencing technology that have transformed the landscape of bacterial genome sequencing. The first-generation, also known as Sanger sequencers, is represented by the ABI Capillary Sequencer (Applied Biosystems). The second-generation, also known as high-throughput sequencers, is represented by MiSeq, a 4-channel sequencer, and NextSeq, a 2-channel sequencer (Illumina), both sequencing by synthesis instruments. Lastly, the third-generation, also known as long-read sequencers, is represented by the Pacific Bioscience BS sequencer and Oxford Nanopore MinION sequencer.
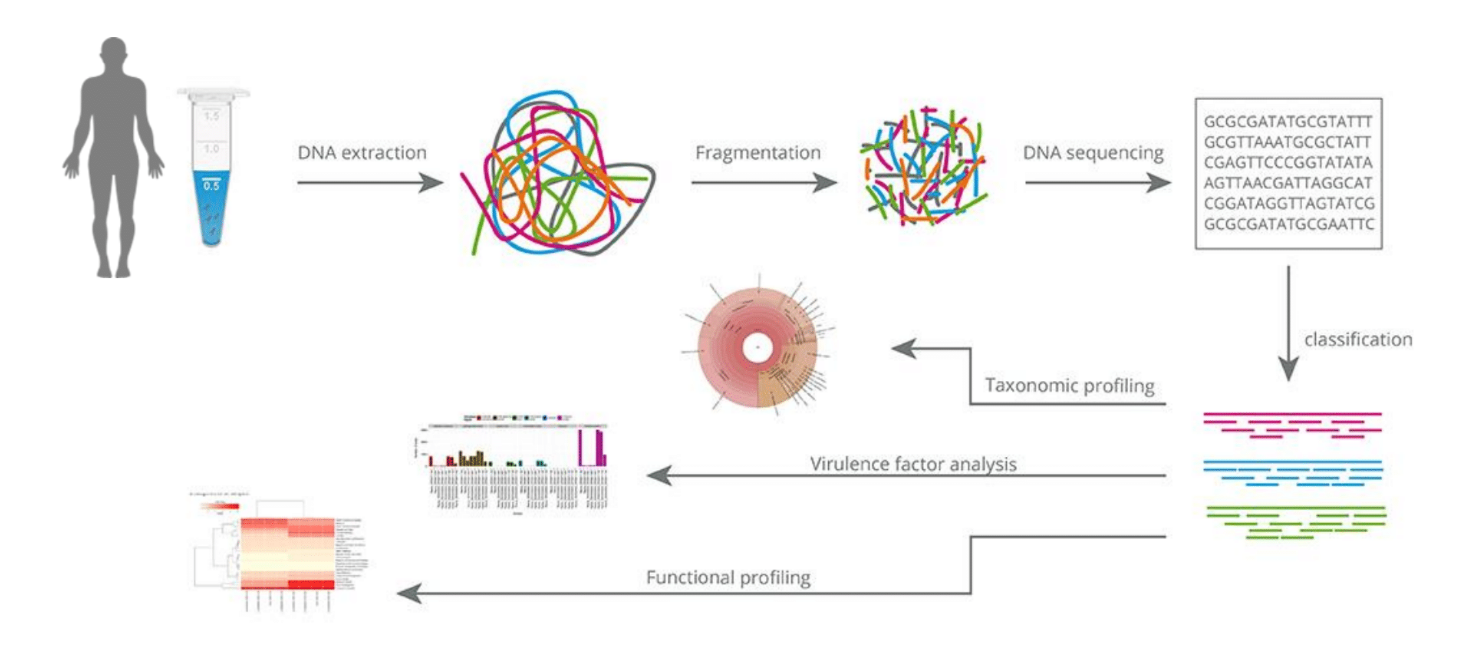
Hypothetical workflow based on metagenomic sequencing. Schematic representation of the hypothetical workflow for the direct processing of samples from suspected sources of pathogens after adoption of metagenomic sequencing, with an expected timescale that could fit within a single day. Adapted from Didelot et al., 2012
https://eurofinsgenomics.eu/en/eurofins-genomics/material-and-methods/metagenome-analysis/
https://www.frontiersin.org/articles/10.3389/fpubh.2022.899077/full
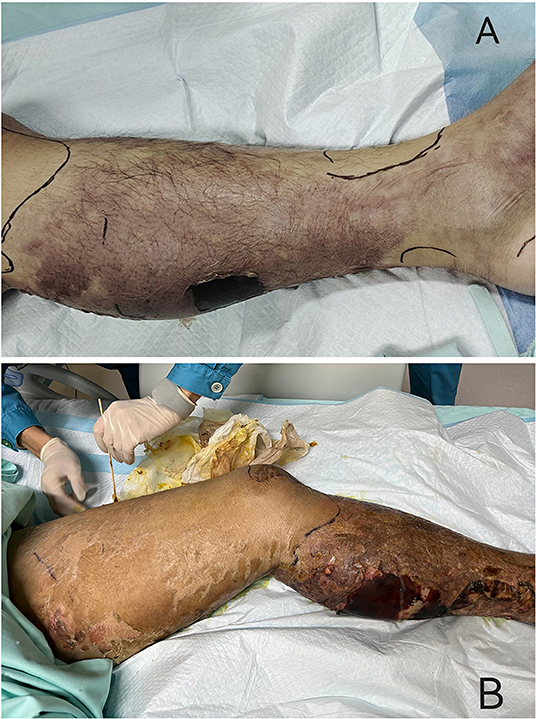
Clinical presentation of the patient. (A) At the admission to ICU, it could be seen the moderate swelling, hemorrhagic blisters and bullae, marked pain with movement, and overlying erythema of his left lower limb. (B) Three days after admission, the area of redness and swelling of the left lower limb gradually spread to the root of the thigh and perineum.
https://www.frontiersin.org/articles/10.3389/fpubh.2022.899077/full
Magic box of NGS Wonders for Microbiology
Completely characterized strain:
Read mapping
Read mapping software
reads
reference genome
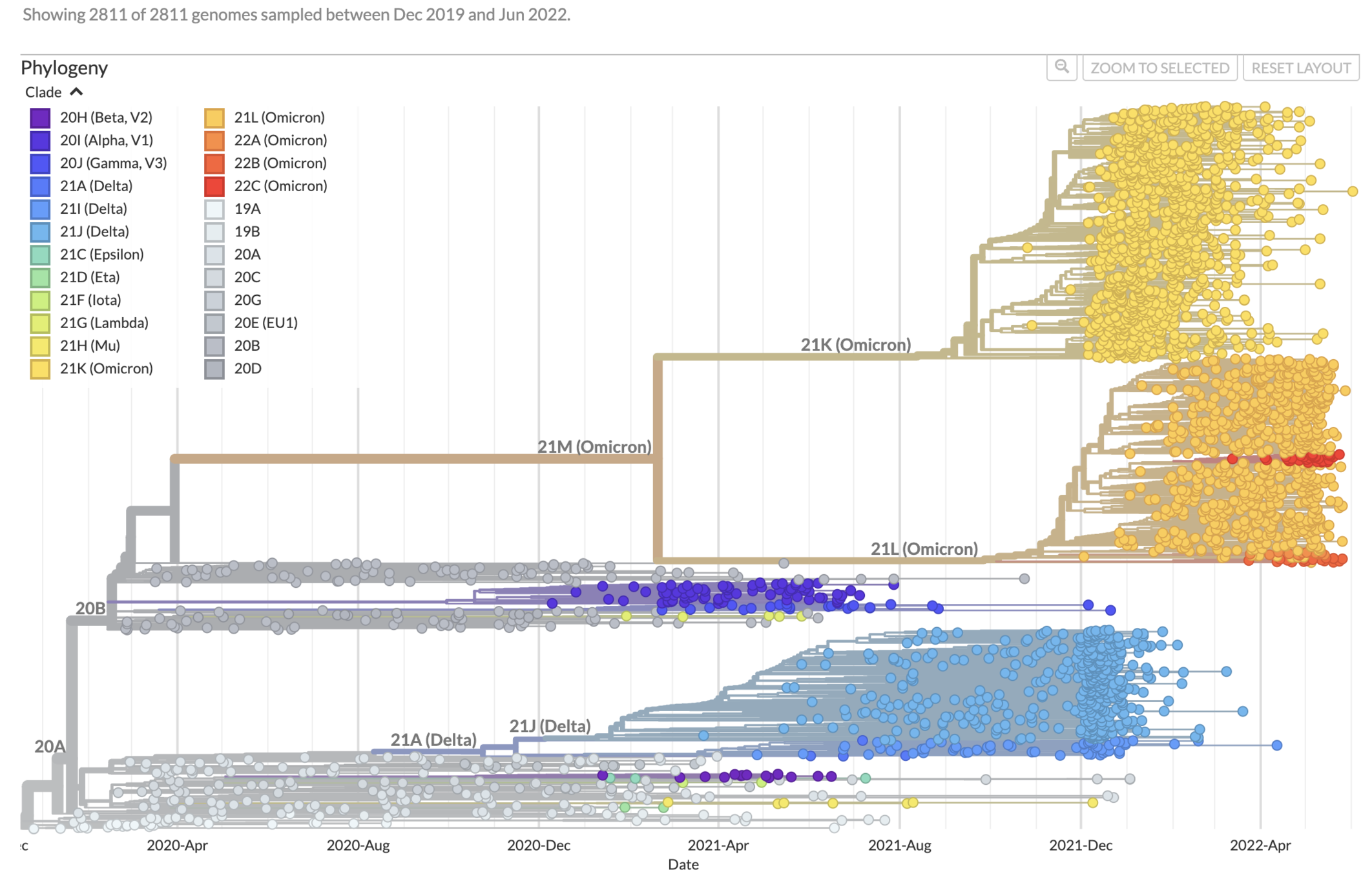
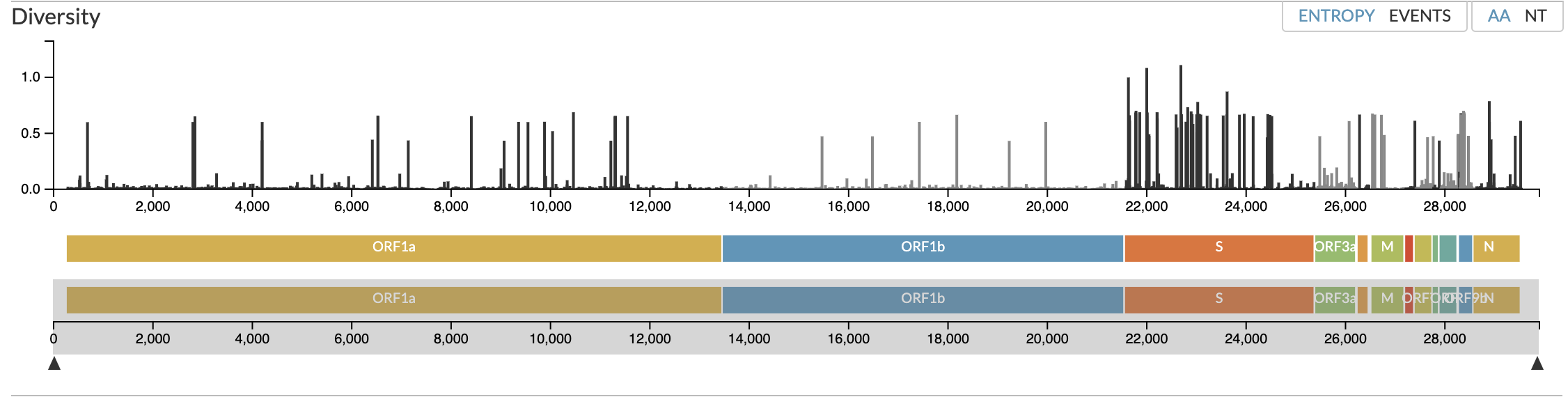
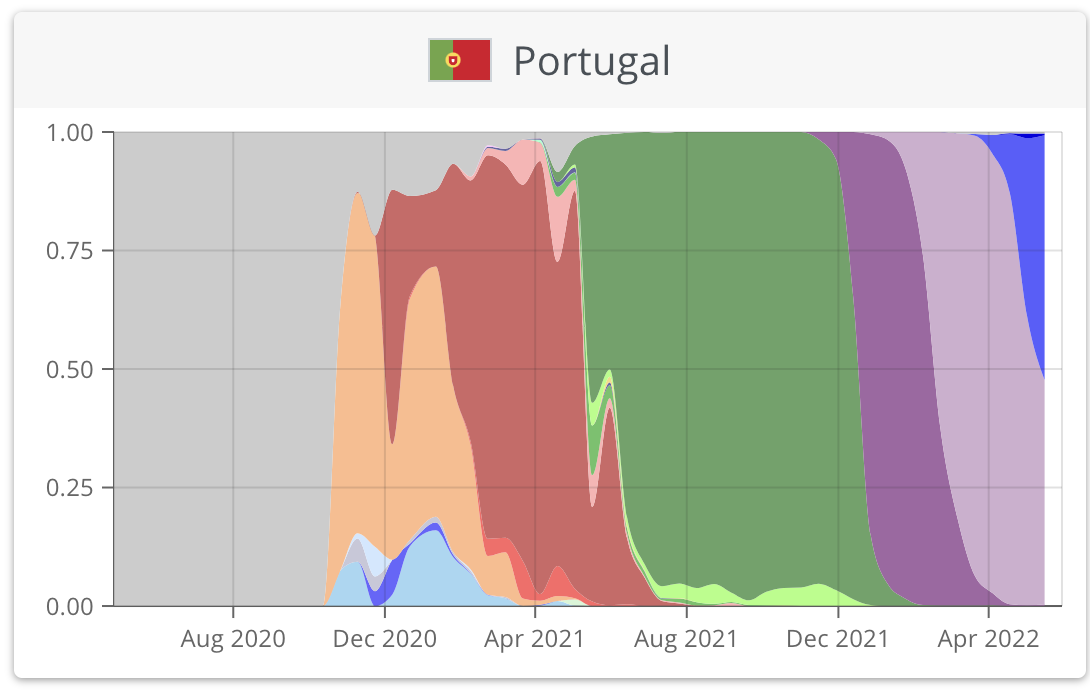
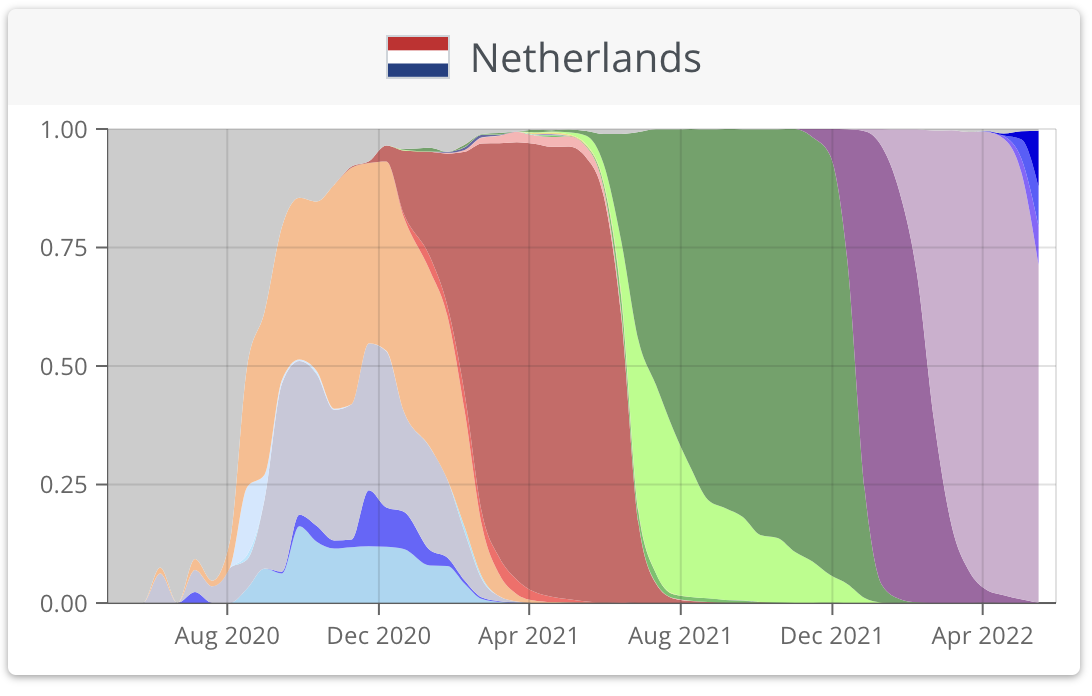
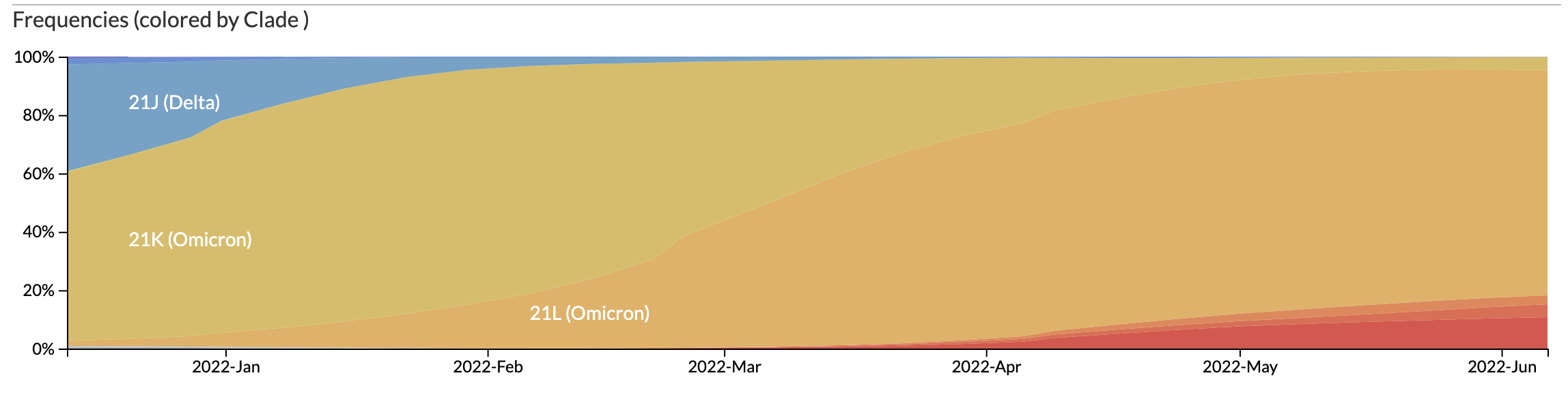
https://nextstrain.org/ncov/gisaid/global/6m
https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---1-june-2022
https://covariants.org/per-country?region=World
https://nextstrain.org/ncov/gisaid/global/6m
Gene-by-Gene
de novo assembly software
reads
contigs
annotation/ comparison
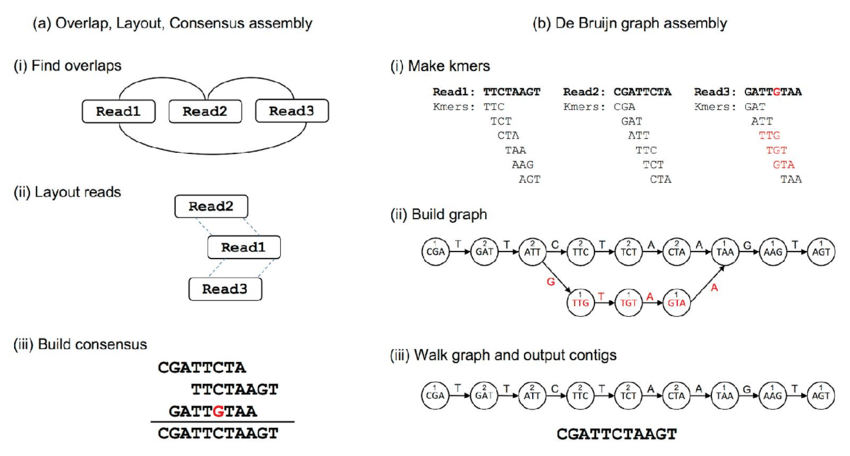
Two different approaches to genome assembly: (a) in Overlap, Layout, Consensus assembly, (i) overlaps are found between reads and an overlap graph constructed (edges indicate overlapping reads). (ii) Reads are laid out into contigs based on the overlaps (dashed lines indicate overlapping portions). (iii) The most likely sequence is chosen to construct consensus sequence. (b) In dBg assembly, (i) reads are decomposed into kmers by sliding a window of size k across the reads. (ii) The kmers become vertices in the dBg, with edges connecting overlapping kmers. Polymorphisms (red) form branches in the graph. A count is kept of how many times a kmer is seen, shown here as numbers above kmers. (iii) Contigs are built by walking the graph from edge nodes. A variety of heuristics handle branches in the graphs—for example, low coverage paths, as shown here, may be ignored.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7299287/
A billion piece puzzle with no reference image
Multilocus Sequence Typing
Why stop here?
Whole-Genome/Core-Genome MLST
| File | Locus 1 | Locus 2 |
|---|---|---|
| genome1.fasta | Allele 1 | Allele 2 |
| genome2.fasta | Allele 1 | Allele 6 |
https://online.phyloviz.net/; https://doi.org/10.1093%2Fnar%2Fgkw359
https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON369
ECDC, EFSA. 2022. Rapid OutbreaK Assessment; Sun et al. 2020. Foodborne Pathog Dis 17:87–97;
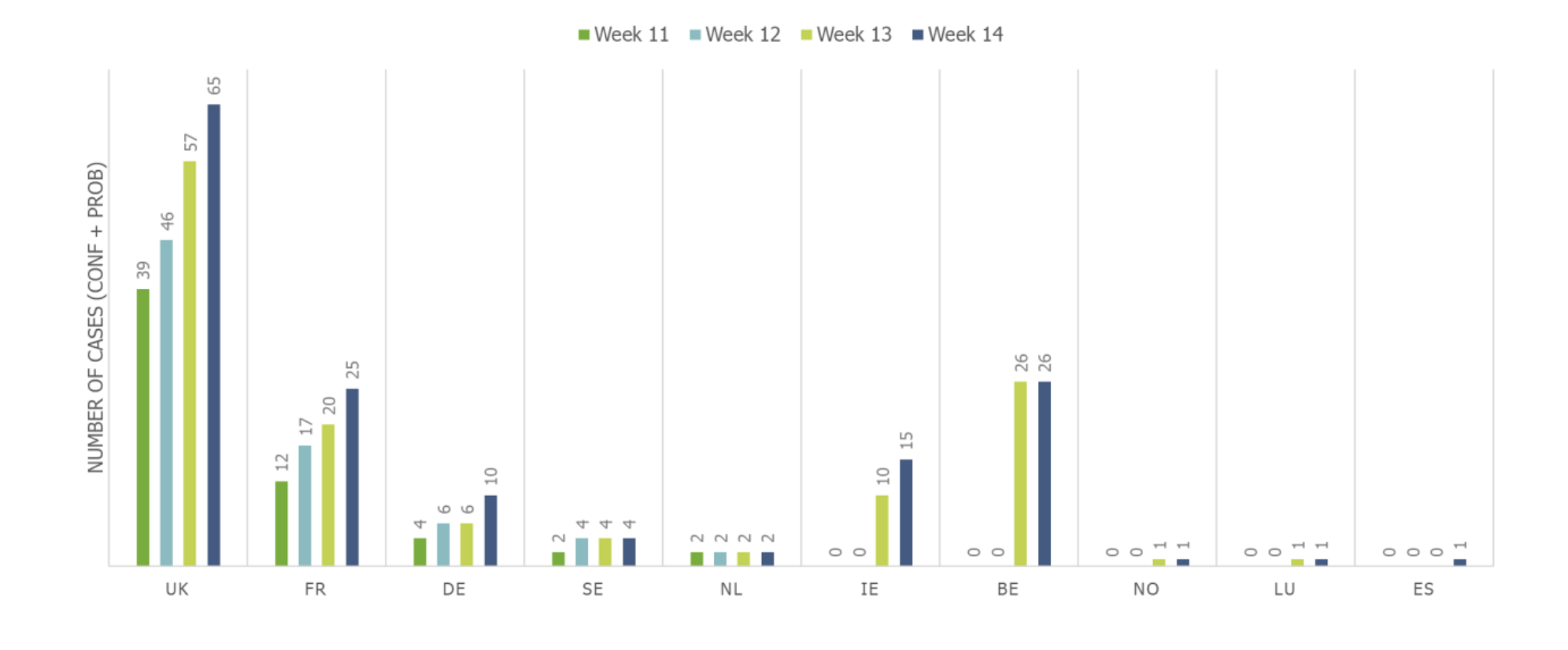
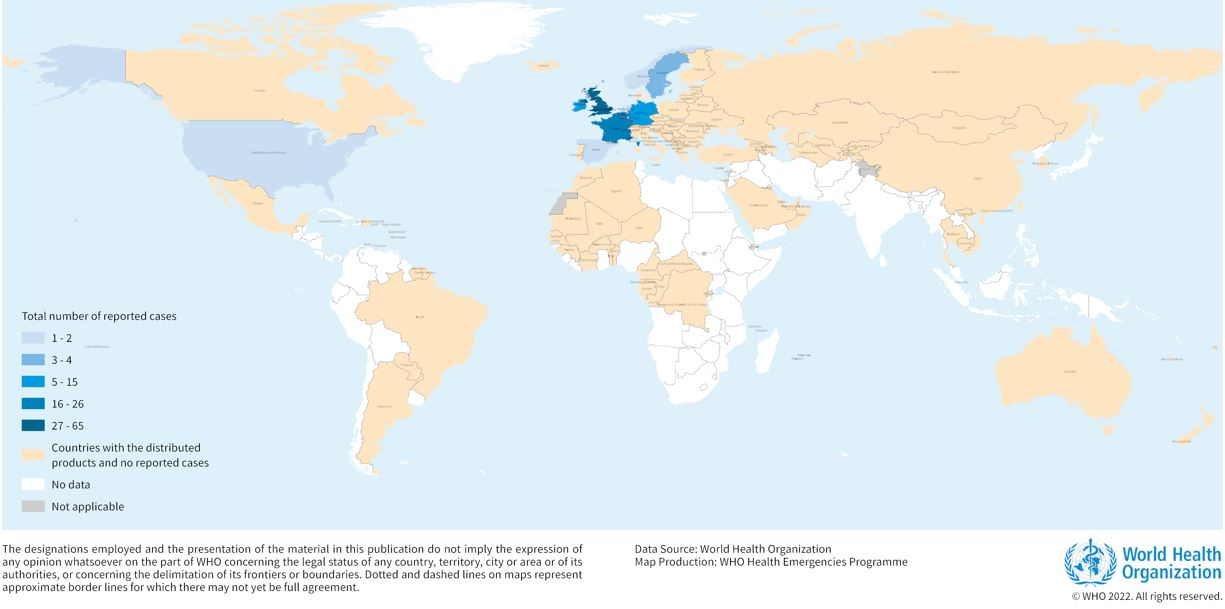
Cumulative number of confirmed and probable monophasic S. Typhimurium cases by week and country in nine EU/EEA countries and the UK, as of 8 April 2022.
Geographical distribution of reported Salmonella Typhimurium outbreak cases (n=151) and countries where implicated products have been distributed (n=113), as of 25 April 2022.
https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON369
ECDC, EFSA. 2022. Rapid OutbreaK Assessment; Sun et al. 2020. Foodborne Pathog Dis 17:87–97;
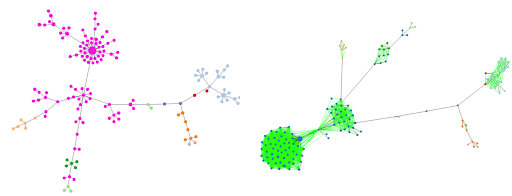
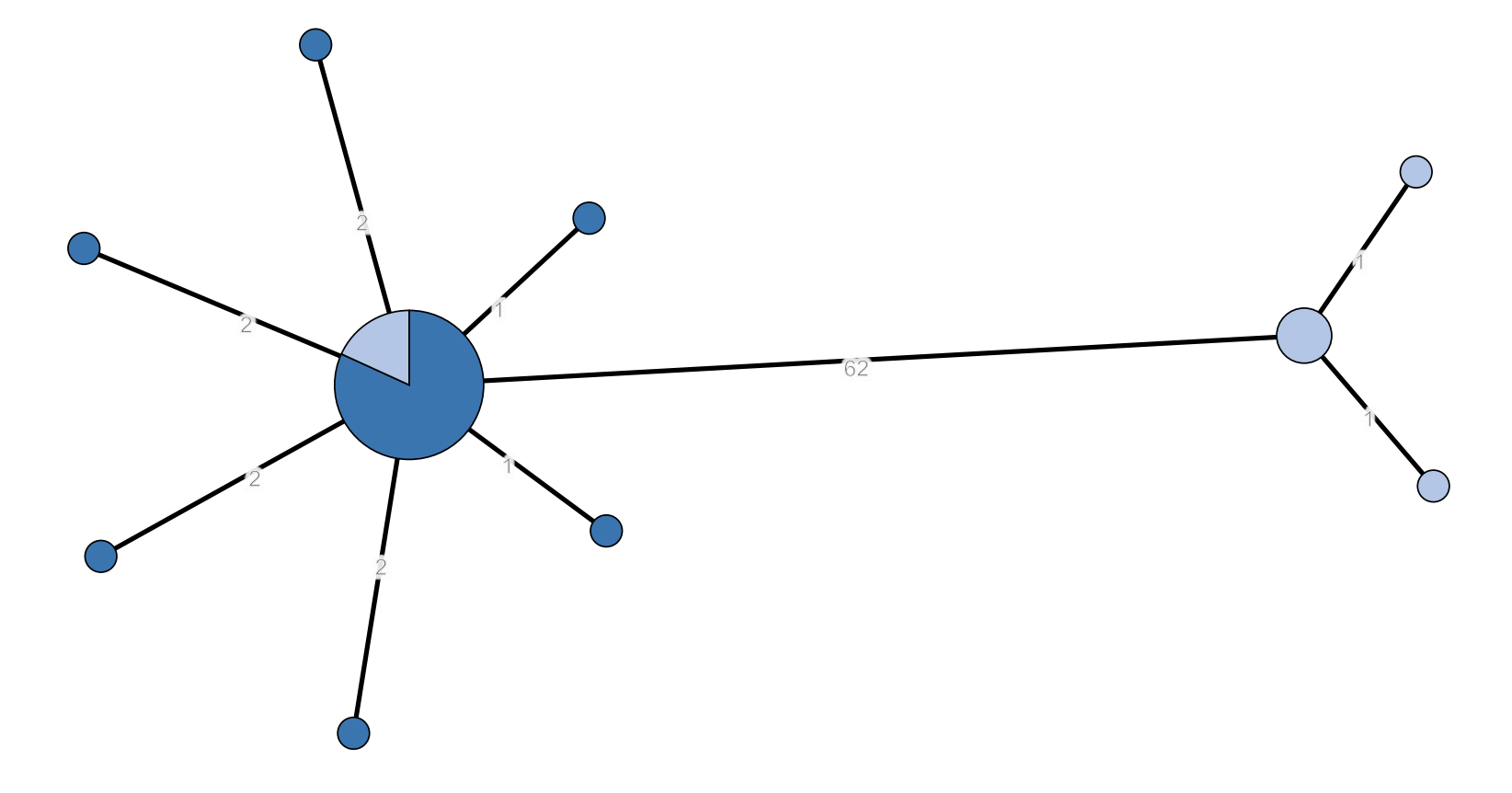
Minimum spanning tree of 24 human (dark blue) and nine non-human (light blue) monophasic Salmonella Typhimurium isolates. cgMLST with 3 225 loci. Cluster includes the representative isolates of the outbreak strain (SRR17830210 and SRR18021617), all human isolates from HC5_296366 and four non-human isolates from the Belgian Processing Plant B.
Soucy, S., Huang, J. & Gogarten, J. Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482 (2015). https://doi.org/10.1038/nrg3962
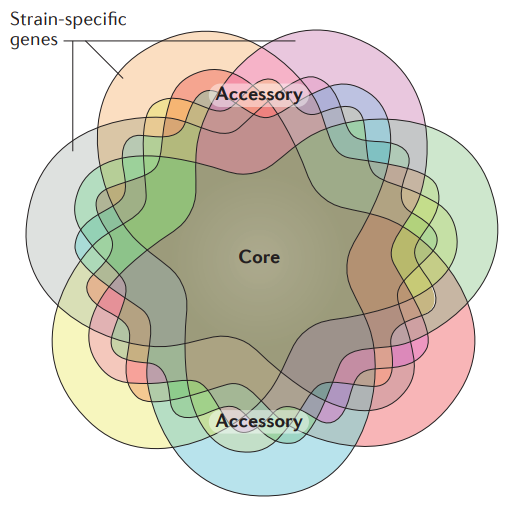
Pangenome:
" (...) hence, core and dispensable genes represent the essence and the diversity of the species, respectively.
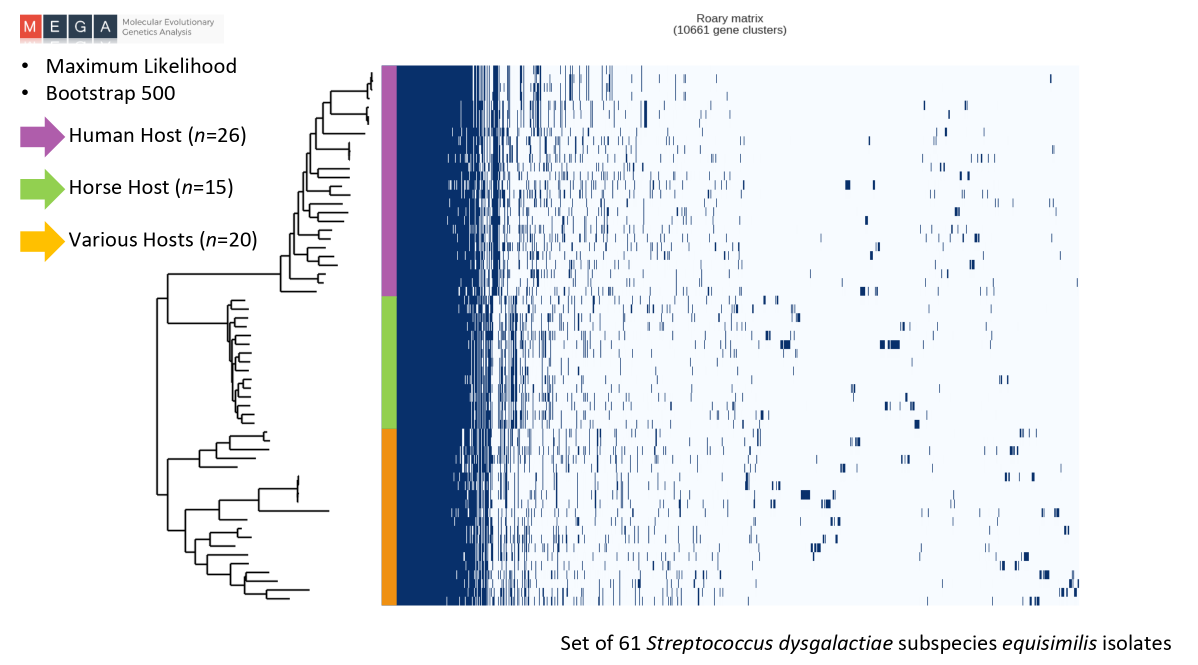
Mendes, CI. Pan-genome comparison between Streptococcus dysgalactiae subsp. equisimilis isolates from human and animal sources. Tese de mestrado, Bioinformática e Biologia Computacional, FCUL, 2016. http://hdl.handle.net/10451/25958
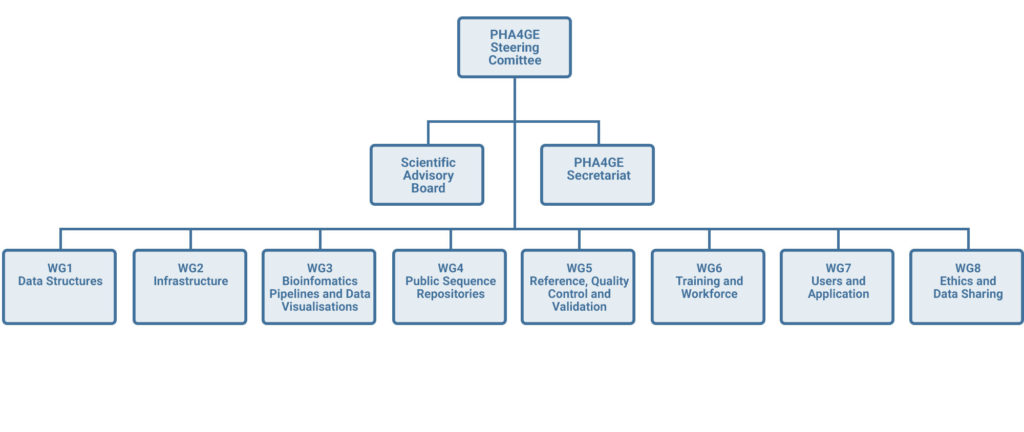
https://pha4ge.org/
Why PHA4GE:
reads
metadata
repository
stakeholders
https://doi.org/10.1093/gigascience/giac003
Algorithms
Data Structures
Interfaces
The role of bioinformatics in clinical microbiology
https://slides.com/
inesmendes/igc_2022
@ines_cim
cimendes
MRamirez Lab, iMM
2019
By Inês Mendes




