






Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
I am on the scientific advisory boards of Apriori Bio, Invivyd, Aerium Therapeutics, the Vaccine, Company, and Oncorus
I am an inventor on Fred Hutch licensed patents related to deep mutational scanning of viral proteins
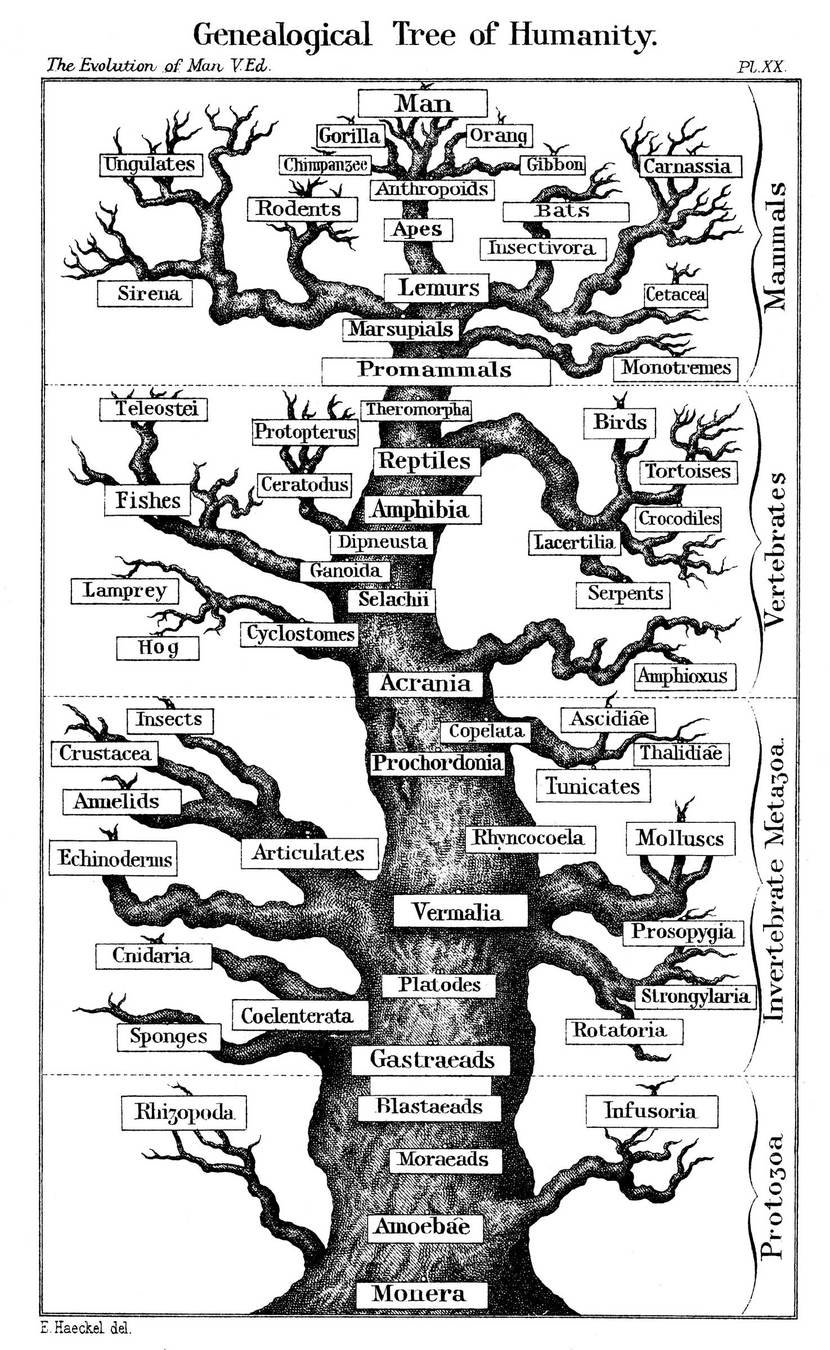
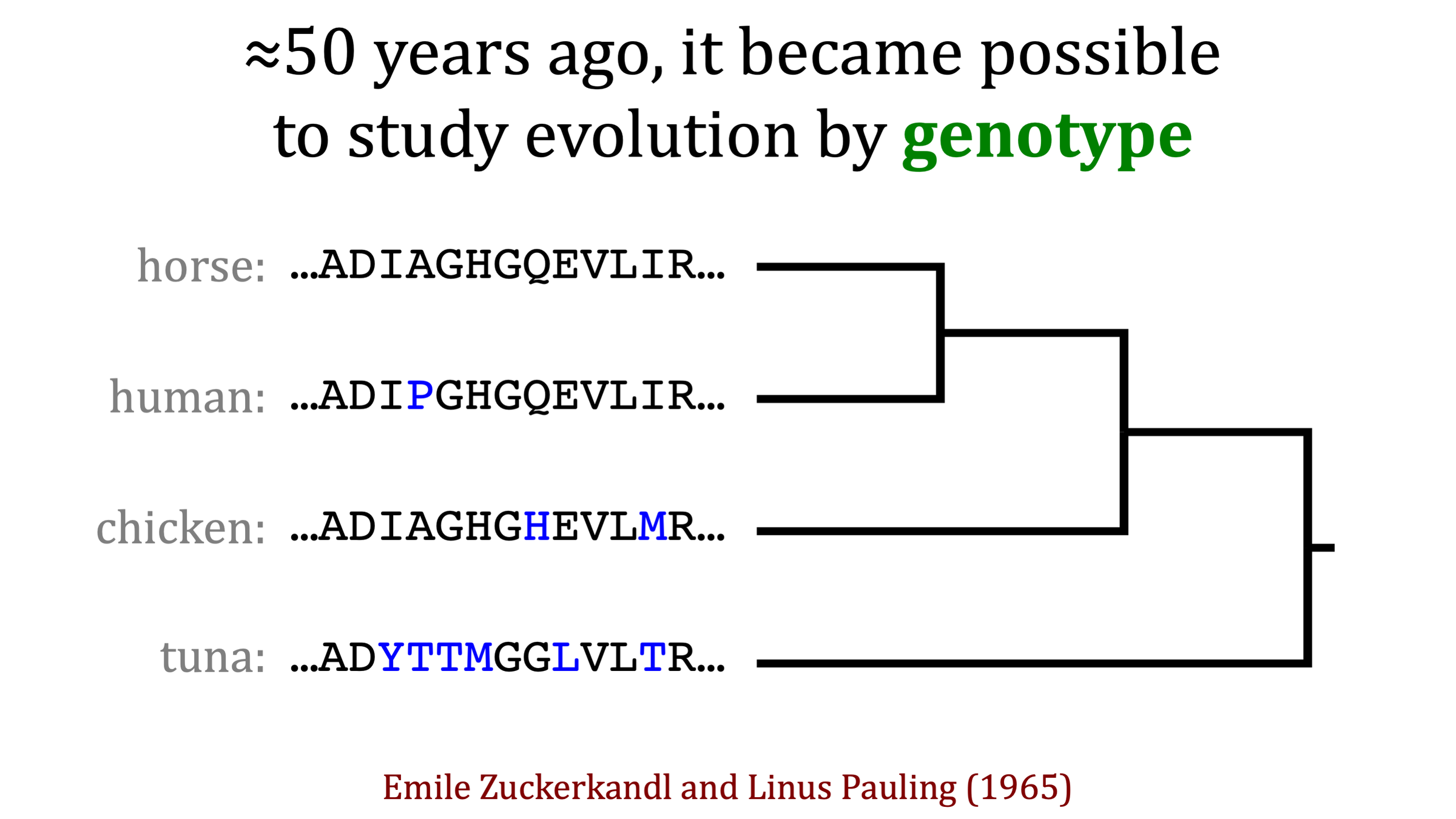
"There is no reason to expect that the extent of functional change in a polypeptide chain is proportional to the number of amino-acid substitutions... It is the type rather than the number of substitutions that is decisive."
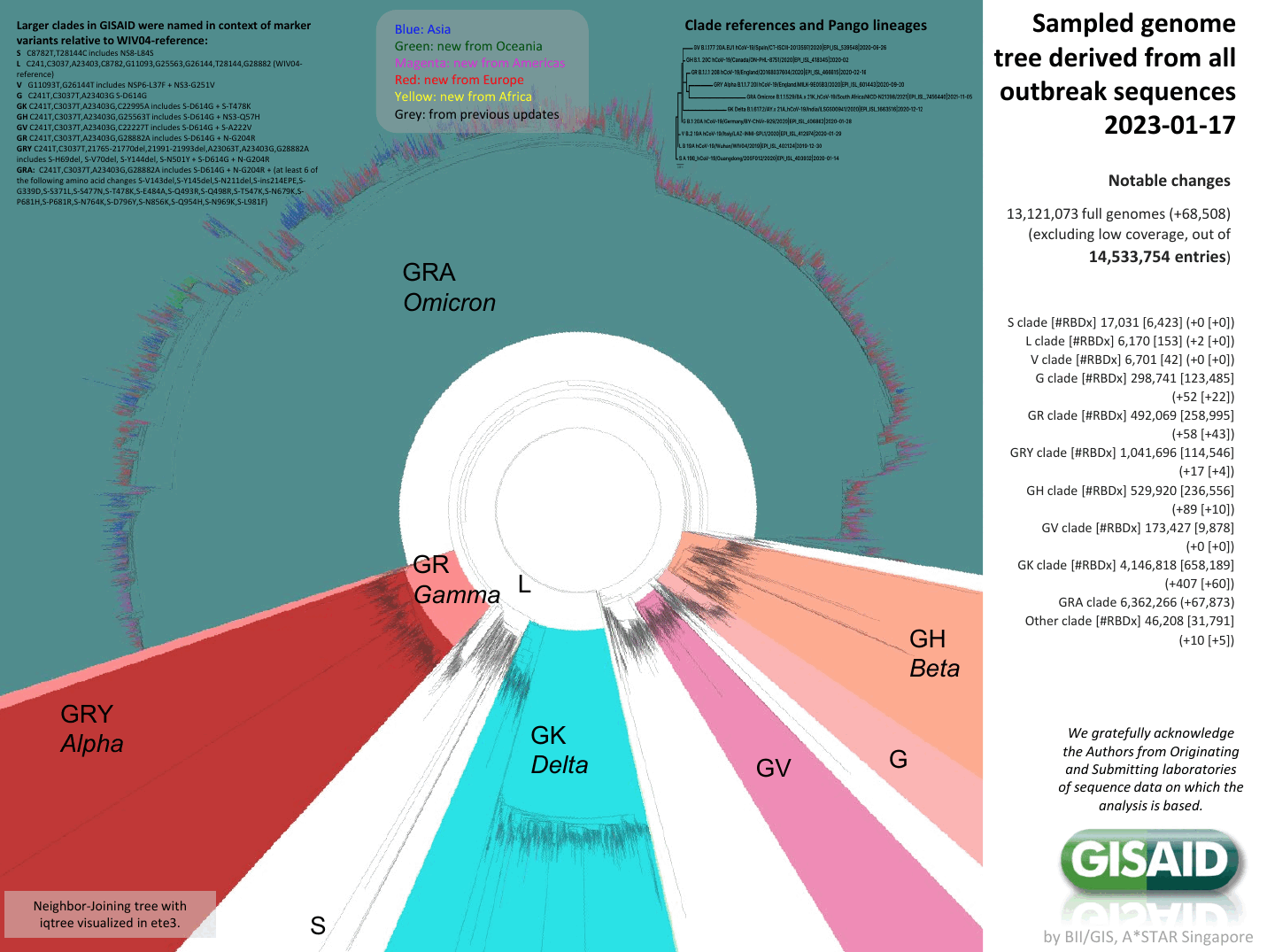
Phylogenetic tree of ~13 million full-length SARS-CoV-2 genomes provided by GISAID
The Coronavirus Is Mutating. What Does That Mean For Us?
The Coronavirus Is Mutating. What Does That Mean For Us?
"There is no reason to expect that the extent of functional change in a polypeptide chain is proportional to the number of amino-acid substitutions... It is the type rather than the number of substitutions that is decisive."
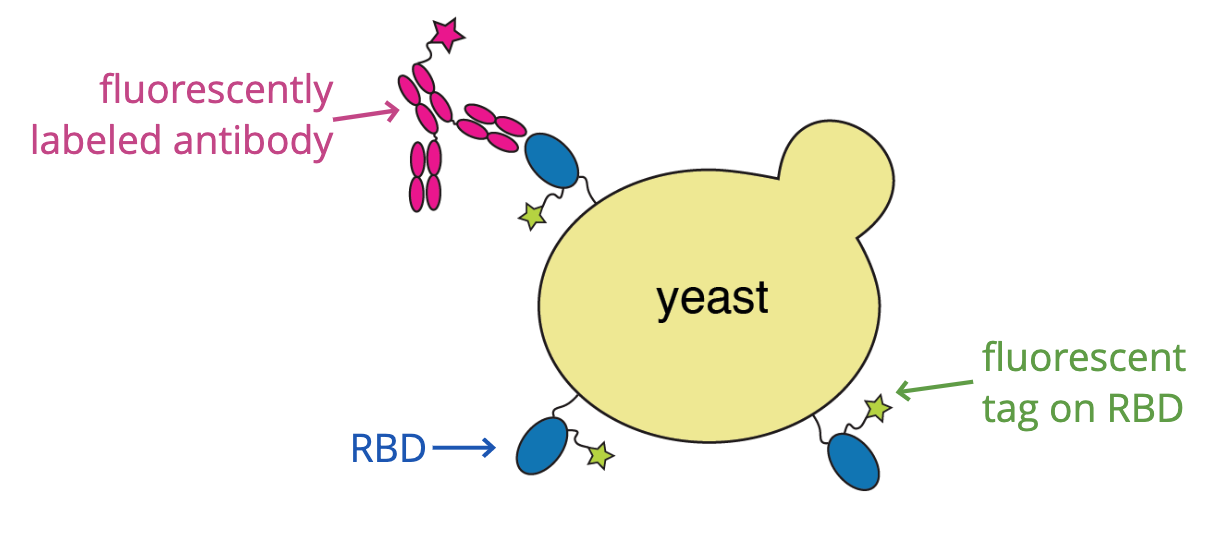
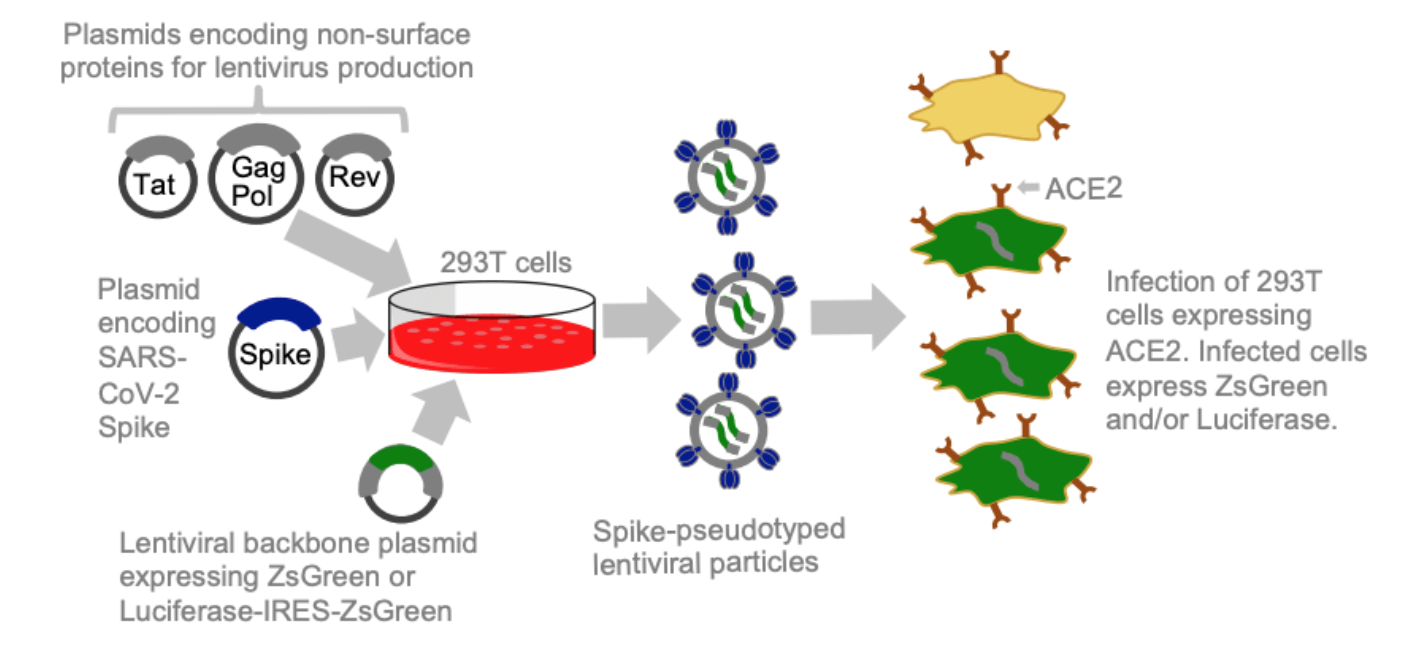
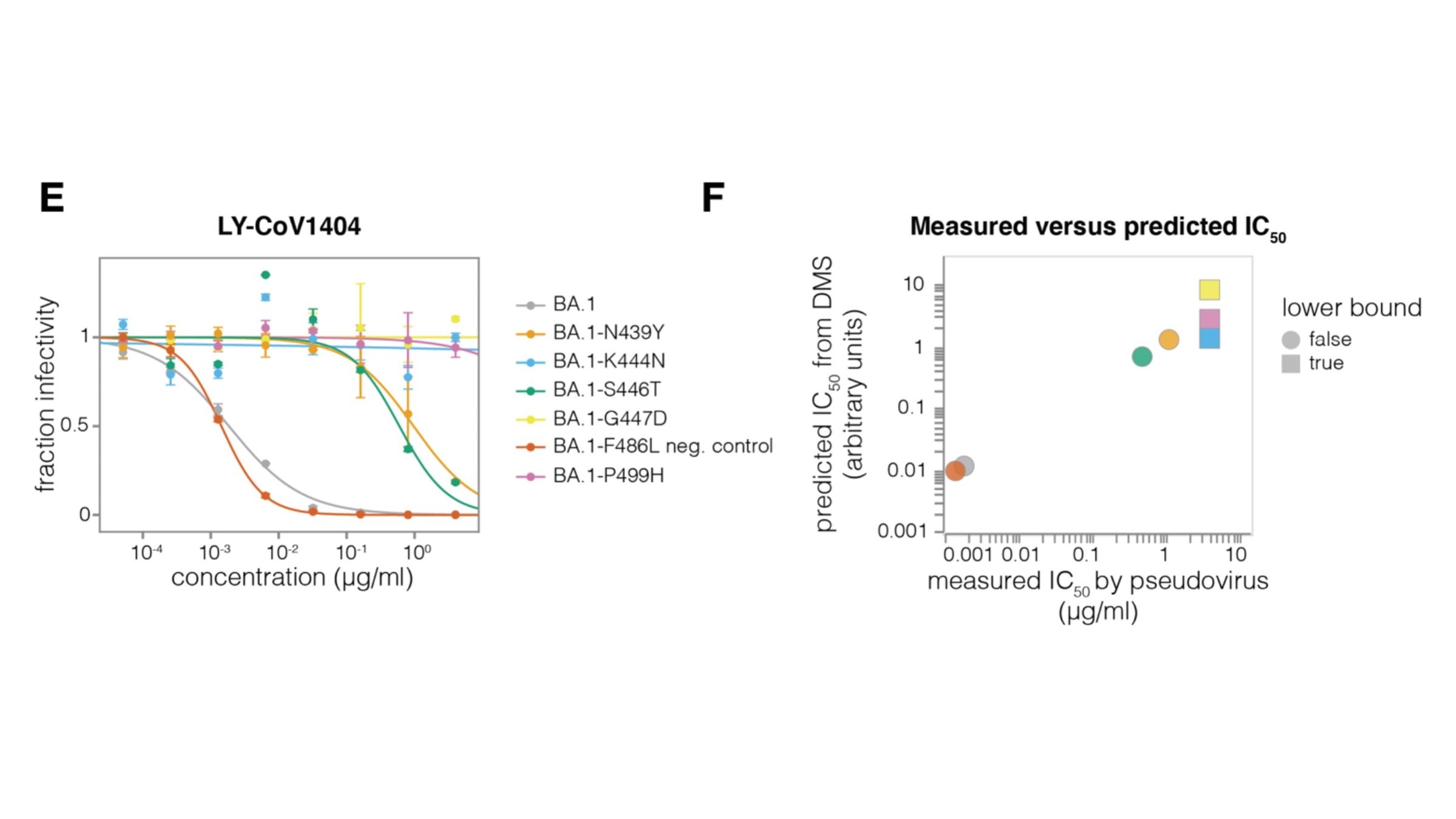
Current deep mutational scanning used by our group and that of Yunlong Cao et al are based on yeast displaying libraries of the spike receptor-binding domain (RBD) and selecting for antibody or ACE2 binding.
Bernadeta Dadonaite
Kate Crawford
Caelan Radford
with Richard Neher
Phylogenetic tree of ~13 million full-length SARS-CoV-2 genomes provided by GISAID
The Coronavirus Is Mutating. What Does That Mean For Us?
"There is no reason to expect that the extent of functional change in a polypeptide chain is proportional to the number of amino-acid substitutions... It is the type rather than the number of substitutions that is decisive."
By Jesse Bloom
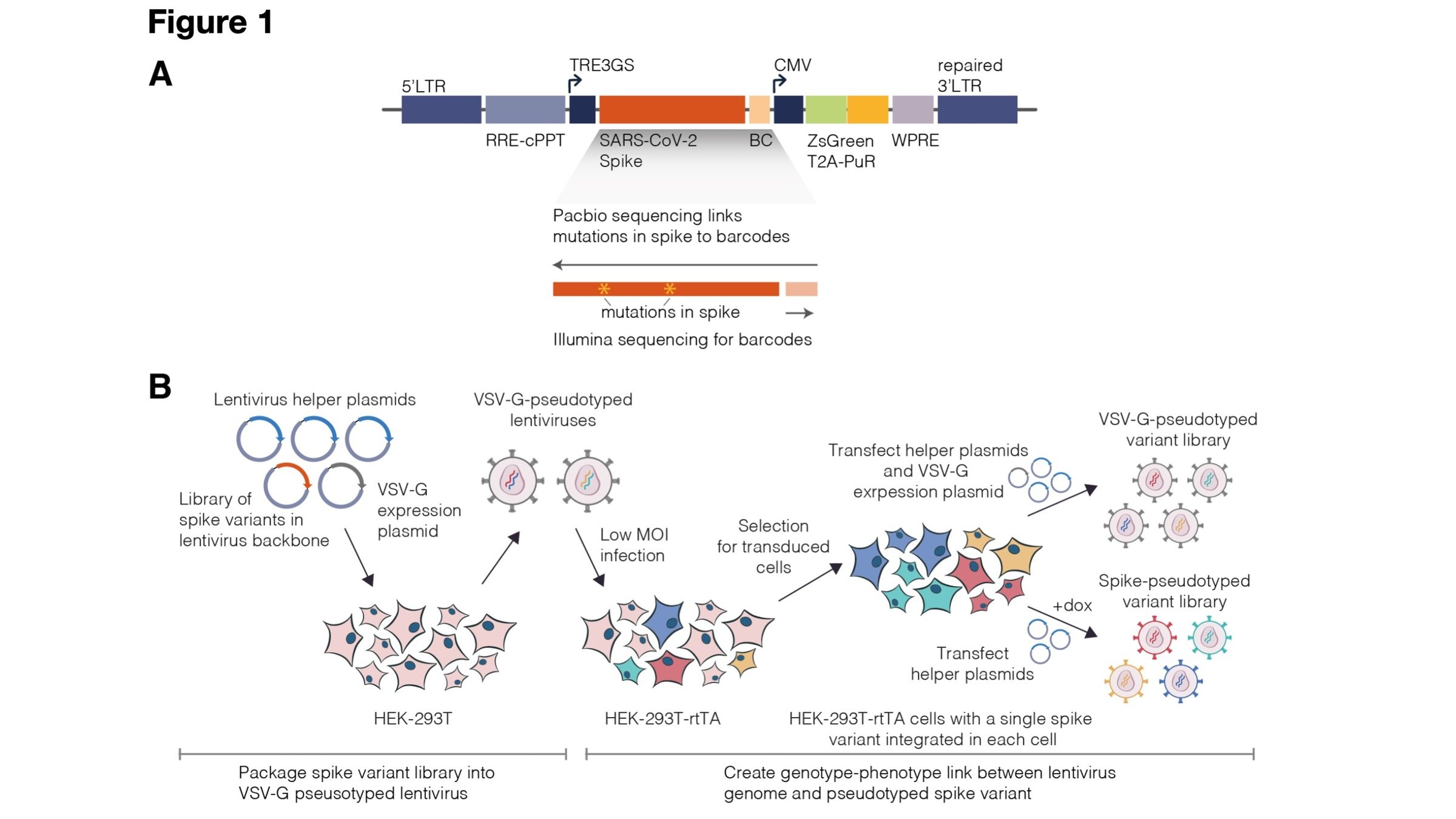
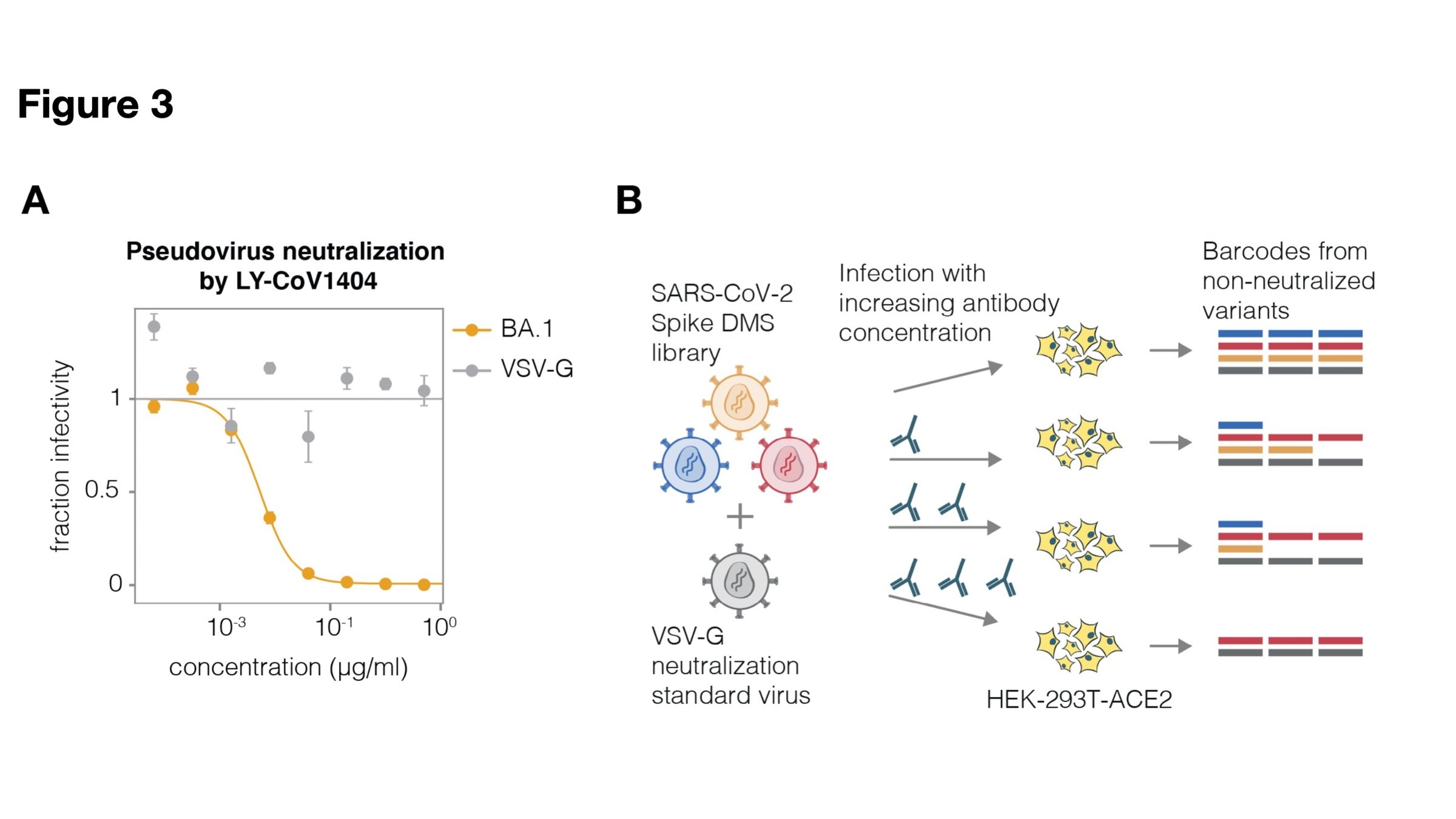
Lentiviral pseudotype deep mutational scanning and estimating mutational effects



