Johannes Köster
https://koesterlab.github.io


The road to reproducibility
-
Snakemake in 2017
dataset
results
Data analysis
"Let me do that by hand..."

dataset
results
dataset
dataset
dataset
dataset
dataset

"Let me do that by hand..."
Data analysis
dataset
results
dataset
dataset
dataset
dataset
dataset
automation
From raw data to final figures:
- document parameters, tools, versions
- execute without manual intervention
Reproducible data analysis
dataset
results
dataset
dataset
dataset
dataset
dataset
scalability
Handle parallelization:
- execute for tens to thousands of datasets
- efficiently use any computing platform
automation
Reproducible data analysis
dataset
results
dataset
dataset
dataset
dataset
dataset
Handle deployment:
be able to easily execute analyses on a different machine
portability
scalability
automation
Reproducible data analysis
dataset
results
dataset
dataset
dataset
dataset
dataset
scalability
automation
portability
dataset
results
dataset
dataset
dataset
dataset
dataset
Define workflows
in terms of rules
Define workflows
in terms of rules
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
script:
"scripts/myscript.R"
rule myfiltration:
input:
"result/{dataset}.txt"
output:
"result/{dataset}.filtered.txt"
shell:
"mycommand {input} > {output}"
rule aggregate:
input:
"results/dataset1.filtered.txt",
"results/dataset2.filtered.txt"
output:
"plots/myplot.pdf"
script:
"scripts/myplot.R"Define workflows
in terms of rules
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
script:
"scripts/myscript.R"
rule myfiltration:
input:
"result/{dataset}.txt"
output:
"result/{dataset}.filtered.txt"
shell:
"mycommand {input} > {output}"
rule aggregate:
input:
"results/dataset1.filtered.txt",
"results/dataset2.filtered.txt"
output:
"plots/myplot.pdf"
script:
"scripts/myplot.R"Define workflows
in terms of rules

Define workflows
in terms of rules
rule mytask:
input:
"data/{sample}.txt"
output:
"result/{sample}.txt"
shell:
"some-tool {input} > {output}"rule name
how to create output from input
The script directive
rule mytask:
input:
"data/{sample}.txt"
output:
"result/{sample}.txt"
script:
"scripts/myscript.py"import pandas as pd
# read table
table = pd.read_table(snakemake.input[0])
# sort values
table.sort_values("somecol", inplace=True)
# write output
table.to_csv(snakemake.output.somename, sep="\t")how to create output from input
# read table
table <- read.table(snakemake@input[[1]])
# sort values
table <- table[order(table$somecol), ]
# write table
write.table(table, file = snakemake@output[["somename"]])how to create output from input
rule mytask:
input:
"data/{sample}.txt"
output:
"result/{sample}.txt"
script:
"scripts/myscript.R"The script directive
from snakemake.remote import S3
s3 = S3.RemoteProvider()
rule mytask:
input:
s3.remote("data/{sample}.txt")
output:
"result/{sample}.txt"
script:
"scripts/myscript.R"Remote files
Remote files
- Amazon S3
- google storage
- (S)FTP
- HTTP(S)
- Dropbox
- WebDav
- XRootD
- GridFTP
- GFAL
- NCBI
Mirror entire I/O to remote
snakemake --default-remote-provider GS --default-remote-prefix mybucketdataset
results
dataset
dataset
dataset
dataset
dataset
scalability
automation
portability
Scheduling
Paradigm:
Workflow definition shall be independent of computing platform and available resources
Rules:
define resource usage (threads, memory, ...)
Scheduler:
- solves multidimensional knapsack problem
- schedules independent jobs in parallel
- passes resource requirements to any backend
Scalable to any platform



workstation
compute server
cluster
grid computing
cloud computing

Command-line interface
# execute workflow locally with 16 CPU cores
snakemake --cores 16
# execute on cluster
snakemake --cluster qsub --jobs 100
# execute in the cloud
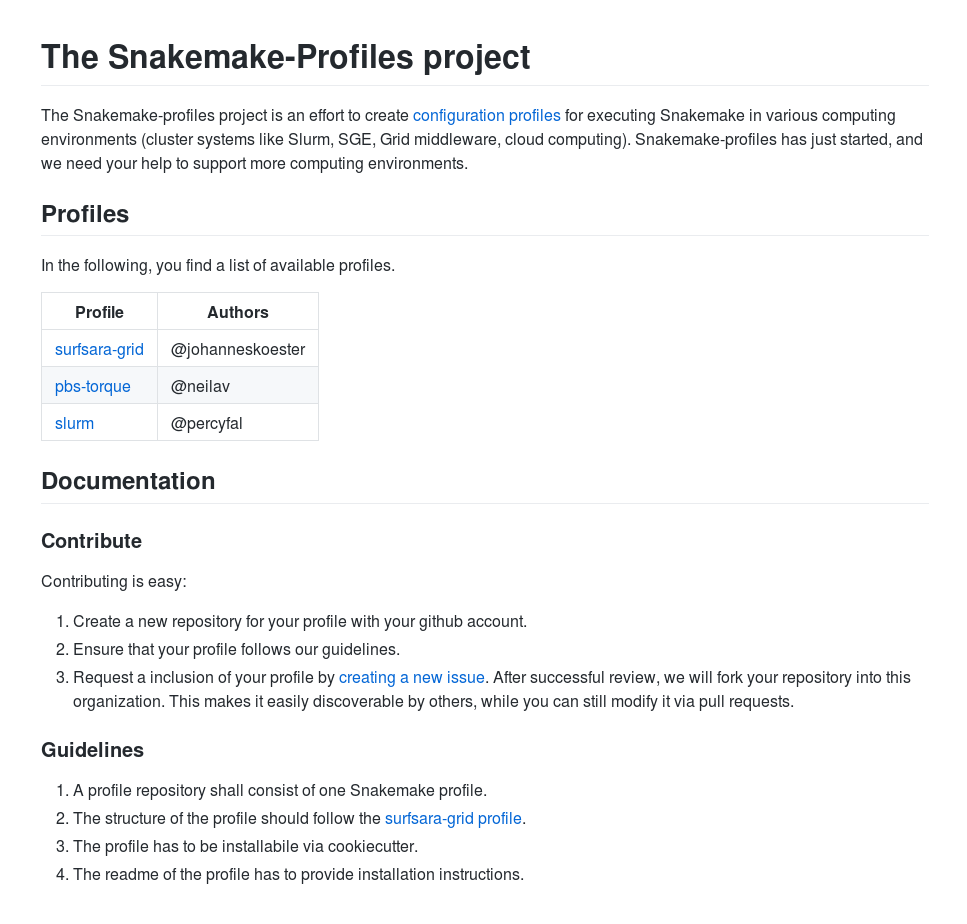
snakemake --kubernetes --jobs 1000 --default-remote-provider GS --default-remote-prefix mybucketConfiguration profiles
snakemake --profile slurm --jobs 1000$HOME/.config/snakemake/slurm
├── config.yaml
├── slurm-jobscript.sh
├── slurm-status.py
└── slurm-submit.pyConfiguration profiles
dataset
results
dataset
dataset
dataset
dataset
dataset
Full reproducibility:
install required software and all dependencies in exact versions
portability
scalability
automation
Software installation is a pain
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2")easy_install snakemake./configure --prefix=/usr/local
make
make installcp lib/amd64/jli/*.so lib
cp lib/amd64/*.so lib
cp * $PREFIXcpan -i bioperlcmake ../../my_project \
-DCMAKE_MODULE_PATH=~/devel/seqan/util/cmake \
-DSEQAN_INCLUDE_PATH=~/devel/seqan/include
make
make installapt-get install bwayum install python-h5pyinstall.packages("matrixpls")Package management with
package:
name: seqtk
version: 1.2
source:
fn: v1.2.tar.gz
url: https://github.com/lh3/seqtk/archive/v1.2.tar.gz
requirements:
build:
- gcc
- zlib
run:
- zlib
about:
home: https://github.com/lh3/seqtk
license: MIT License
summary: Seqtk is a fast and lightweight tool for processing sequences
test:
commands:
- seqtk seqIdea:
Normalization installation via recipes
#!/bin/bash
export C_INCLUDE_PATH=${PREFIX}/include
export LIBRARY_PATH=${PREFIX}/lib
make all
mkdir -p $PREFIX/bin
cp seqtk $PREFIX/bin- source or binary
- recipe and build script
- package
Easy installation and management:
no admin rights needed
conda install pandas
conda update pandas
conda remove pandas
conda env create -f myenv.yaml -n myenvIsolated environments:
channels:
- conda-forge
- defaults
dependencies:
- pandas ==0.20.3
- statsmodels ==0.8.0
- r-dplyr ==0.7.0
- r-base ==3.4.1
- python ==3.6.0Package management with
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
conda:
"envs/mycommand.yaml"
shell:
"mycommand {input} > {output}"Integration with Snakemake
channels:
- conda-forge
- defaults
dependencies:
- mycommand ==2.3.1Over 3000 bioinformatics related packages


Over 200 contributors






Singularity
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
singularity:
"docker://biocontainers/mycommand"
shell:
"mycommand {input} > {output}"Coming soon:
Singularity + Conda
rule mytask:
input:
"path/to/{dataset}.txt"
output:
"result/{dataset}.txt"
singularity:
"docker://ubuntu:16.04"
conda:
"envs/mycommand.yaml"
shell:
"mycommand {input} > {output}"Snakemake Wrappers
rule star:
input:
sample=["reads/{sample}.1.fastq", "reads/{sample}.2.fastq"]
output:
"star/{sample}/Aligned.out.bam"
log:
"logs/star/{sample}.log"
params:
# path to STAR reference genome index
index="index",
# optional parameters
extra=""
threads: 8
wrapper:
"0.19.3/bio/star/align"
- deploy software via Conda
- obtain script from git repository
Snakemake Wrappers
import os
from snakemake.shell import shell
extra = snakemake.params.get("extra", "")
log = snakemake.log_fmt_shell(stdout=True, stderr=True)
n = len(snakemake.input.sample)
assert n == 1 or n == 2, "input->sample must have 1 (single-end) or 2 (paired-end) elements."
if snakemake.input.sample[0].endswith(".gz"):
readcmd = "--readFilesCommand zcat"
else:
readcmd = ""
outprefix = os.path.dirname(snakemake.output[0]) + "/"
shell(
"STAR "
"{snakemake.params.extra} "
"--runThreadN {snakemake.threads} "
"--genomeDir {snakemake.params.index} "
"--readFilesIn {snakemake.input.sample} "
"{readcmd} "
"--outSAMtype BAM Unsorted "
"--outFileNamePrefix {outprefix} "
"--outStd Log "
"{log}")Snakemake Wrappers
Snakemake Workflows

Sustainable publishing
Goal:
- persistently store reproducible data analysis
- ensure it is always accessible
Minimize dependencies:
- third-party resources
- proprietary formats
Solution:
- store everything (including packages) in an archive
- upload to persistent storage like https://zenodo.org
- obtain document object identifier (DOI)
Sustainable publishing
# archive workflow (including Conda packages)
snakemake --archive myworkflow.tar.gzAuthor:
- Upload to Zenodo and acquire DOI.
- Cite DOI in paper.
Reader:
- Download and unpack workflow archive from DOI.
# execute workflow (Conda packages are deployed automatically)
snakemake --use-conda --cores 16Conclusion
- Fully reproducible data analyses have to consider three dimensions.
- In the last months, Snakemake has seen improvements in all three dimensions of reproducibility.
- The result is an almost complete and future-proof solution to one of the major scientific challenges of today.
portability
scalability
automation
The road to reproducibility - Snakemake in 2017
By Johannes Köster
The road to reproducibility - Snakemake in 2017
Keynote at Snakemake Day 2017 in Amsterdam (https://snakemake-days.github.iohttps://snakemake-days.github.io



