ANEMIAS DECORRENTES DE DOENÇAS DA MEDULA ÓSSEA
CAPÍTULO 13, 14, 15 E 16
TRATADO DE HEMATOLOGIA ZAGO
ANEMIAS DECORRENTES DE DOENÇAS DA MEDULA ÓSSEA
CLASSIFICAÇÕES:
ANEMIA APLÁSTICA
HEMOGLOBINÚRIA PAROXÍSTICA NOTURNA
ANEMIA DE FANCONI
ANEMIA APLÁSTICA
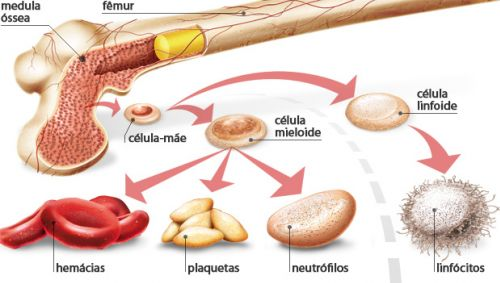
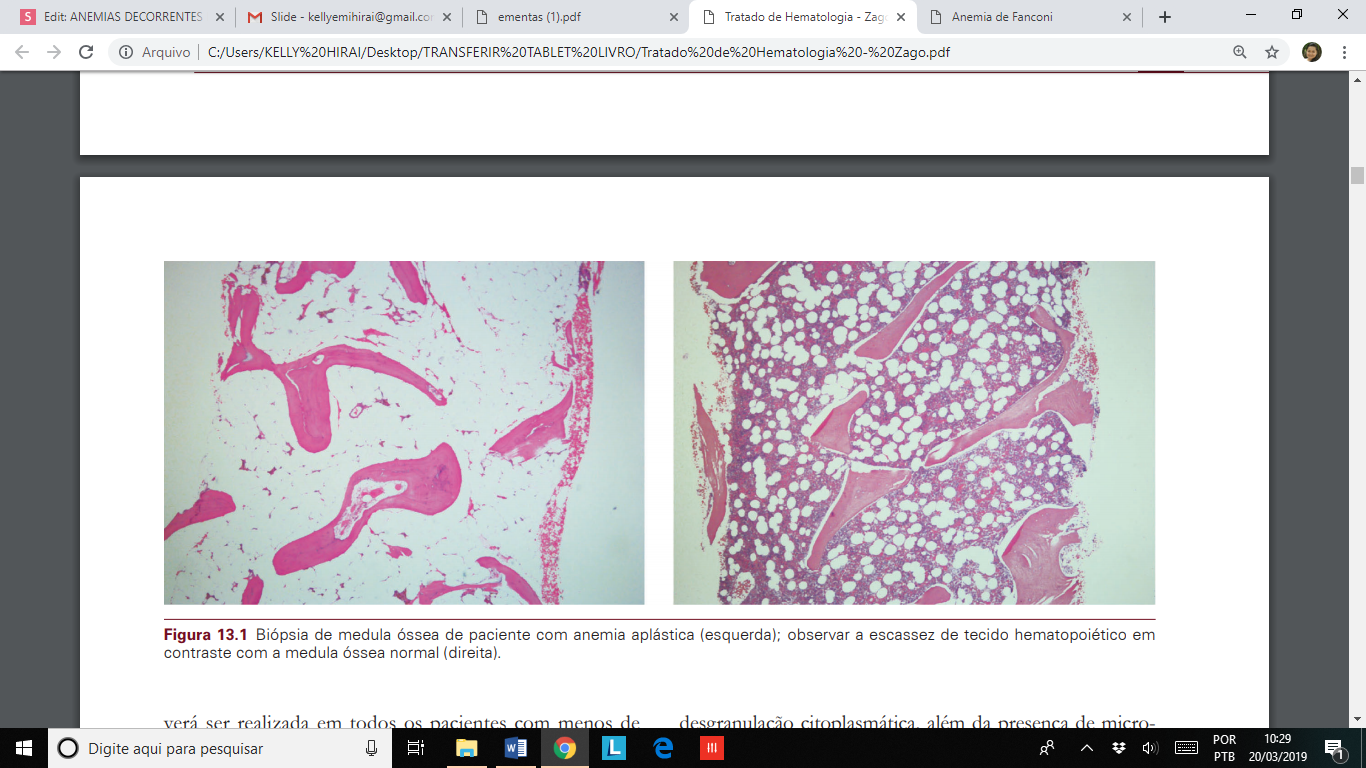
Anemia aplástica é uma entidade rara e heterogênea, caracterizada por pancitopenia no sangue periférico, associada à medula óssea hipocelular, e sem evidência de infiltração neoplásica, mieloproliferativa ou fibrose. Por definição, a biópsia de medula será intensamente hipocelular, substituída por gordura, e no mielograma serão vistos escassos linfócitos, plasmócitos e fibroblastos.
ETIOLOGIA:
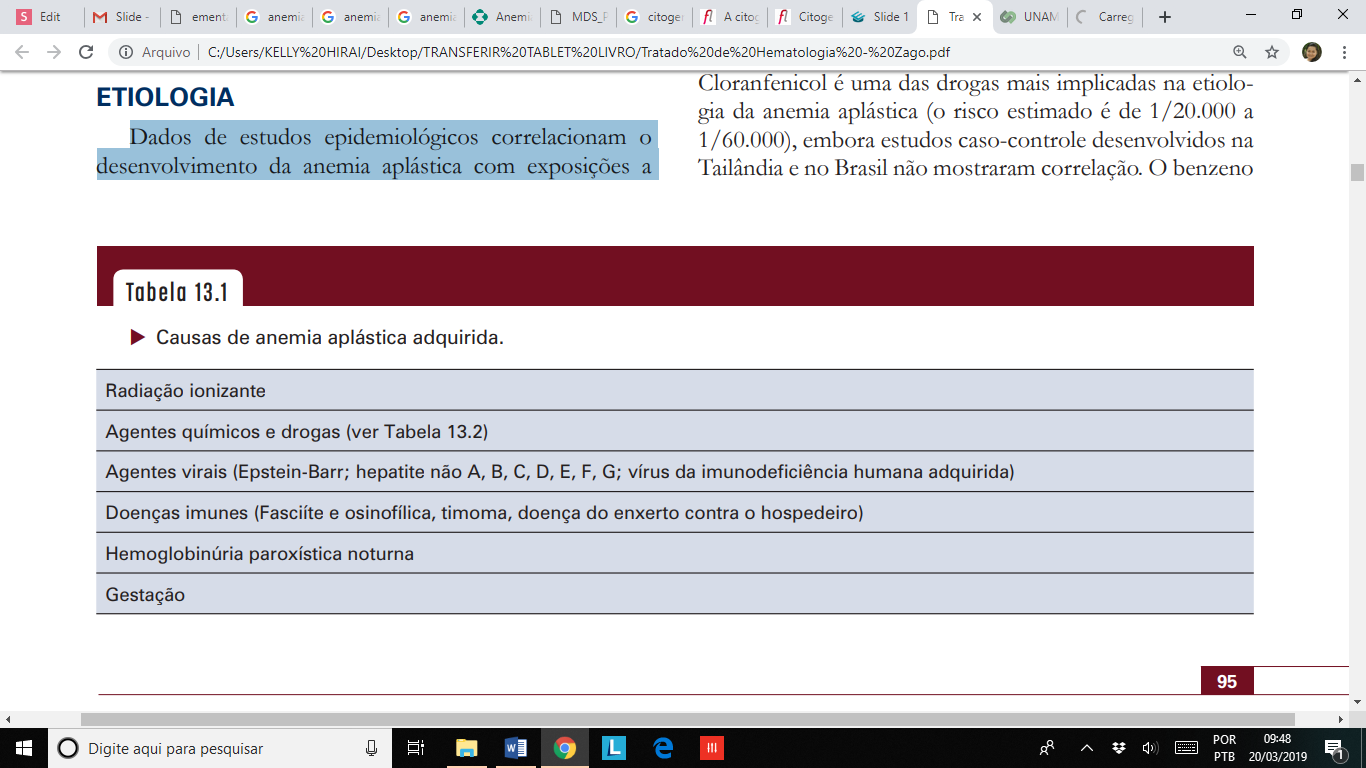
Dados de estudos epidemiológicos correlacionam o desenvolvimento da anemia aplástica com exposições a drogas, agentes químicos, radiação e a uma variedade de doenças . Em 60 a 75% dos casos, não há evidência de um agente causal, sendo então denominada de anemia aplástica idiopática.
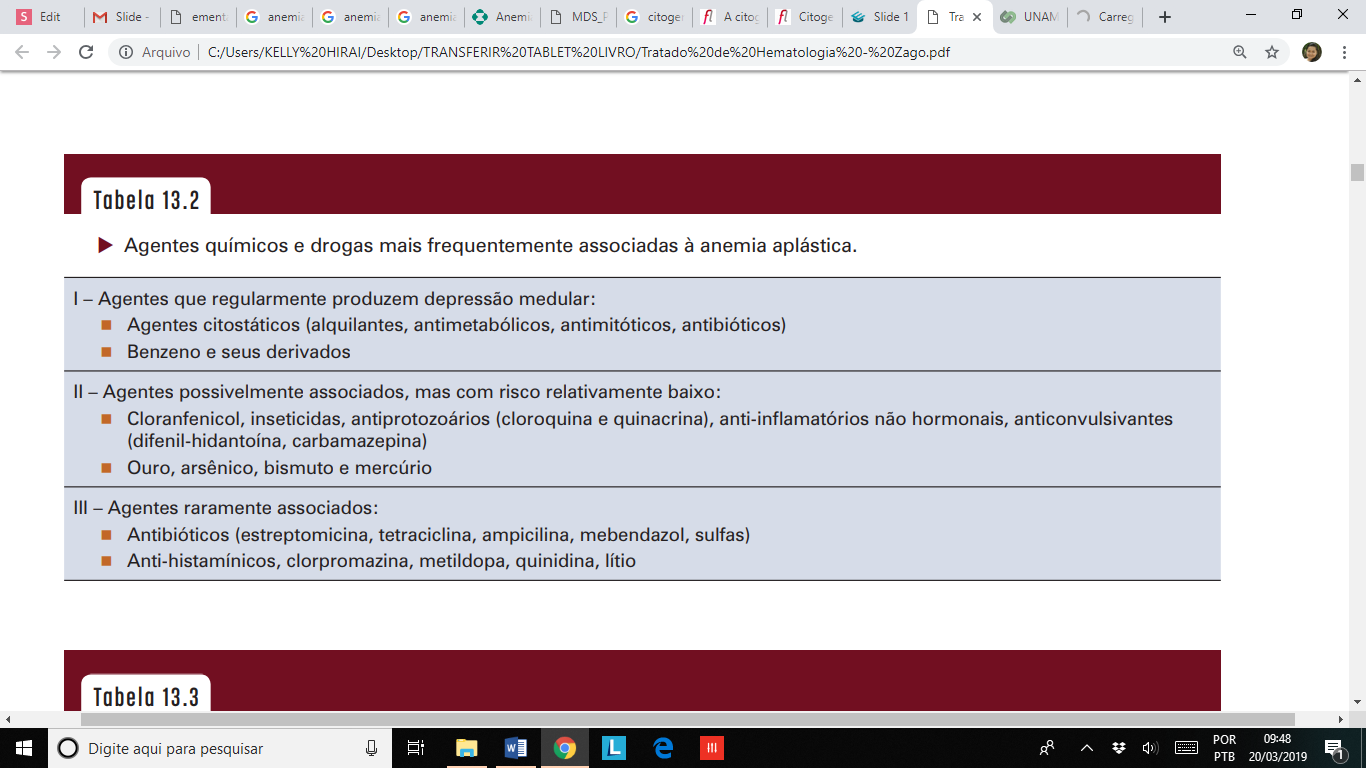
Os mecanismos responsáveis pelo desenvolvimento da anemia aplástica adquirida não são totalmente conhecidos, e incluem:
1) lesão intrínseca da célula progenitora hematopoética;
2) participação imune no desencadeamento e manutenção das citopenias;
3) perturbações do microambiente da medula óssea; e
4) mutações no gene da telomerase e encurtamento telomérico.
FISIOPATOLOGIA
CLASSIFICAÇÃO:
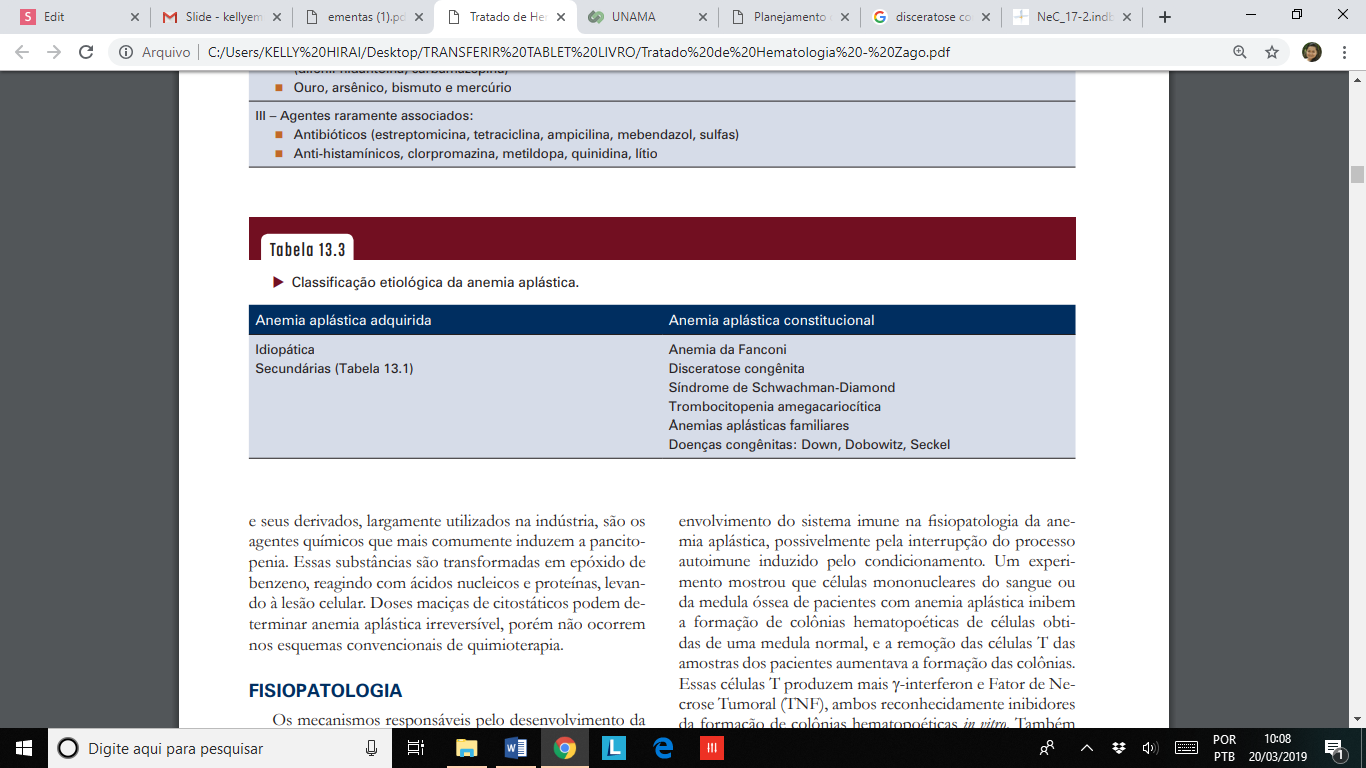
A anemia aplástica pode ser adquirida ou constitucional. É considerada adquirida quando não há qualquer fator predisponente para o seu desenvolvimento e constitucional quando há associação a determinadas doenças congênitas, genéticas ou familiares.
ACHADOS LABORATORIAIS
A pancitopenia é o achado invariável na anemia aplástica, podendo o número absoluto de linfócitos ser normal. O diagnóstico deve ser questionado se as três séries hematopoéticas não estiverem diminuídas.
ACHADOS LABORATORIAIS
As hemácias são normocrômicas e moderadamente macrocíticas, com reticulocitopenia.
A neutropenia absoluta é de importância prognóstica.
Monocitopenia é comum e a produção de linfócitos pode estar normal.
Plaquetas estão, invariavelmente, diminuídas e são qualitativamente normais.
ACHADOS LABORATORIAIS
Ferritina plasmática estará aumentada no início, devido à baixa utilização do ferro, e os pacientes maciçamente transfundidos terão níveis de ferritina muito elevados.
A produção de eritropoetina também estará aumentada, resultante do estímulo induzido pela anemia persistente.
CASO CLÍNICO
Identificação: Homem, 22 anos, solteiro, natural de Grandes Rios (PR), procedente de São Carlos (SP), digitador.
Queixa: Fraqueza e cansaço há 3 meses.
Há três meses apresentou dispnéia e cansaço aos esforços físicos moderados. Há 2 meses e meio apresentou dois episódios de epistaxe necessitando tamponamento anterior em serviço básico de saúde. Este quadro piorou gradativamente passando a apresentar dispnéia e cansaço aos mínimos esforços e epistaxe freqüentes. Procurou serviço médico em sua cidade sendo diagnosticado plaquetopenia; foi tratado com corticoesteróide (prednisona 60 mg/dia com redução lenta da dose), Tagamet, vitamina B12 e ácido fólico. Há 1 semana apresentou febre contínua (38ºC) acompanhada de diarréia, a qual evoluiu para enterorragia. Foi internado em sua cidade por quatro dias recebendo 6 transfusões sanguíneas sem melhora importante do quadro.
EXAMES LABORATORIAIS:
Hemograma:
GB 1.600 (VN: 3.500-10-500)
Hb 6.1 (VN: 12-15)
Ht 17.3% (VN: 35-45%)
VCM 87.8 (VN: 75-92)
HCM 31.0 (VN: 27-32)
Plaquetas 7.000 (VN: 150.000-450.000)
Diferencial com 16% de neutrófilos segmentados (VN: 47-78%), 82% de linfócitos (VN: 16-43%) e 2% de monócitos (VN: 7%).
Reticulócitos 0.1% (VN 1-2,25%)
VHS=56mm (1 h) (VN <10mm).
Mielograma: medula óssea obtida por punção esternal, acentuadamente hipocelular. Série eritrocitária hipocelular, com diseritropoese leve sem alterações megaloblásticas; série branca hipocelular constituída por 87% de linfócitos maduros, 13% neutrófilos segmentados e raros mastócitos.
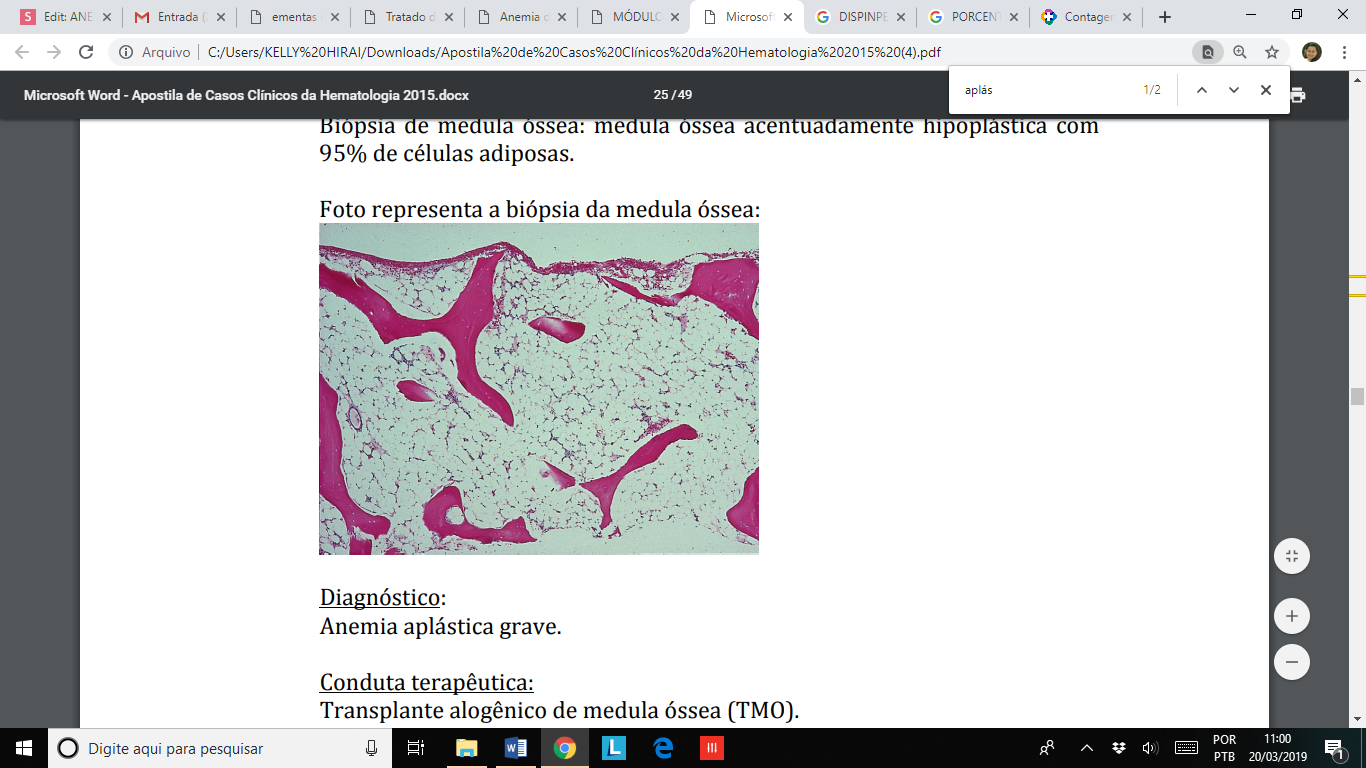
Biópsia de medula óssea: medula óssea acentuadamente hipoplástica com 95% de células adiposas.
Foto representa a biópsia da medula óssea:
Hemoglobinúria Paroxística Noturna
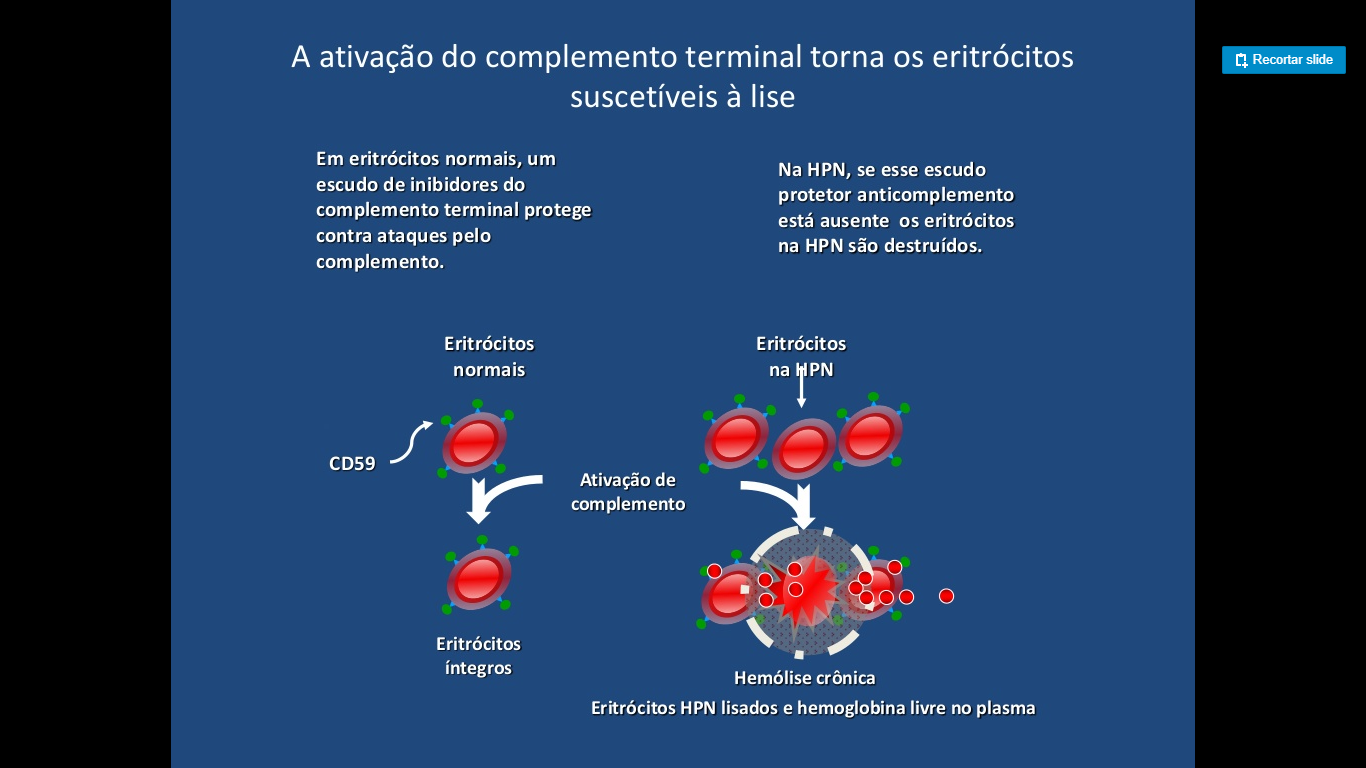
A Hemoglobinúria Paroxística Noturna (HPN) é uma doença clonal da Célula-Tronco Hematopoética (CTH), que resulta na produção de células sanguíneas exibindo alterações características.
É uma anemia hemolítica, crônica adquirida e rara. Seu curso clínico é extremamente variável.
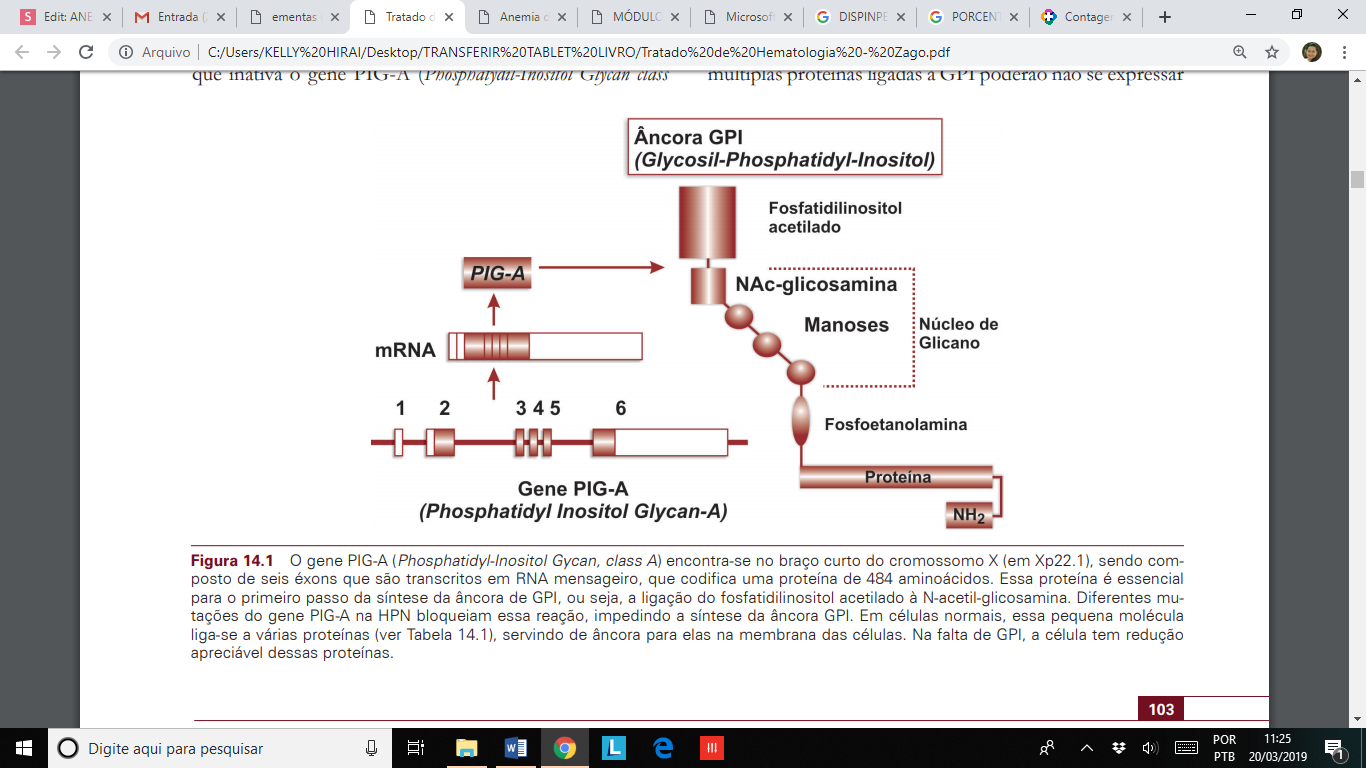
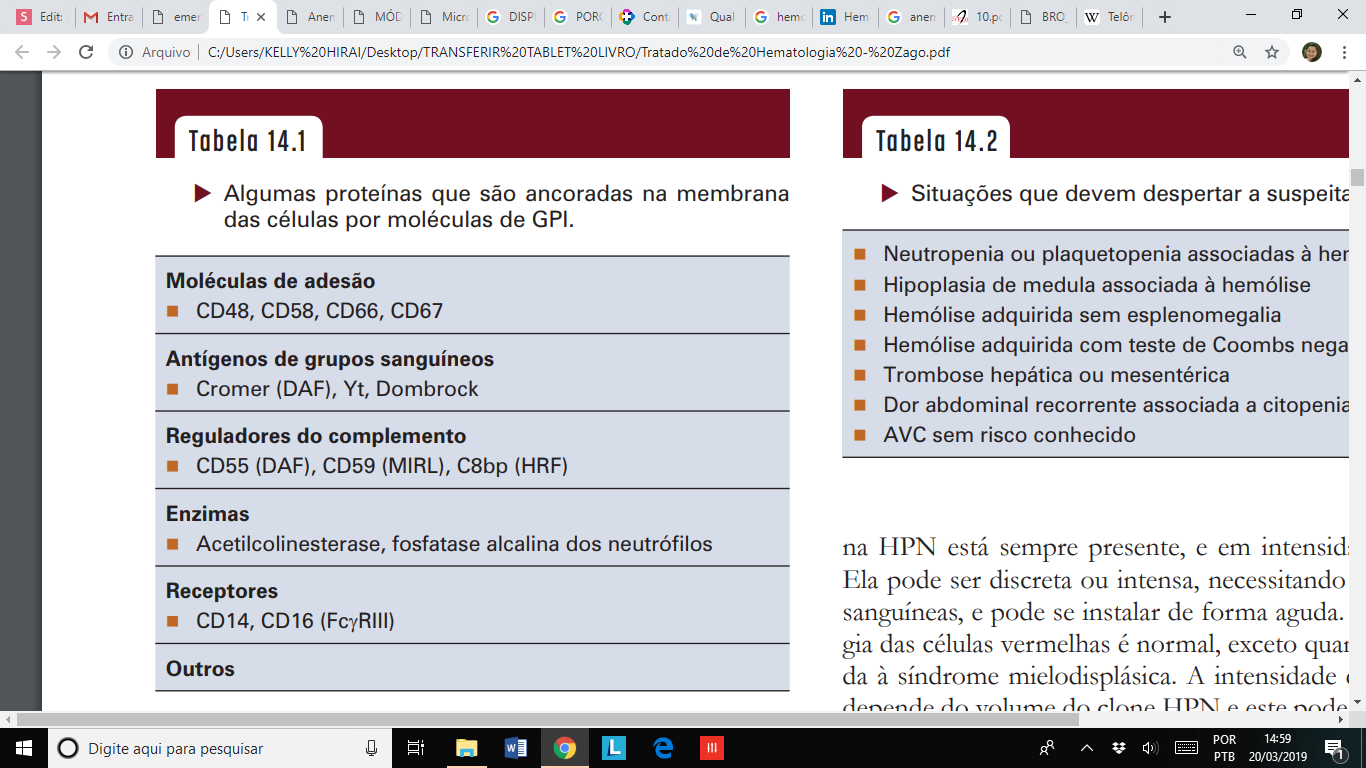
O CD55 (ou decay accelerating factor) inibe o complemento no nível de C3, acelerando a taxa de destruição de C3-convertase ligada à membrana e reduzindo a quantidade de C3 dissociada a C3a e C3b.
Já o CD59 (ou membrane inhibitor of reactive lysis) é uma glicoproteína que interage diretamente com o complexo de ataque à membrana (também chamado de complexo de complemento terminal - C5b9) para impedir a formação do poro lítico na superfície celular a partir da agregação de C9.
A HPN é uma doença hematológica adquirida e rara, apresenta três características clínicas básicas, com expressão variada: hemólise intravascular, tendência à trombose e insuficiência da medula óssea.
Apresenta-se frequentemente com infecções recorrentes, neutropenia, trombocitopenia



DIAGNÓSTICO
Hemograma
Reticulócitos
Desidrogenase lática (LDH)
Bilirrubina
Coombs direto negativo
Teste de Ham (O teste de Ham fundamenta-se na ativação do complemento pela acidificação do soro a um pH de 6,2, condições em que as células HPN sofrem hemólise e as normais não)
Citometria de fluxo para CD55 e CD59
ANEMIA DE FANCONI
Anemia de Fanconi (AF) é uma doença rara, geralmente herdada de maneira autossômica recessiva, e caracterizada por insuficiência medular progressiva, anormalidades congênitas e grande predisposição ao desenvolvimento de mielodisplasia, leucemias e tumores sólidos de cabeça e pescoço.
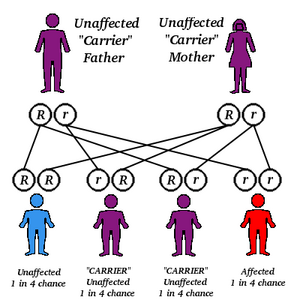
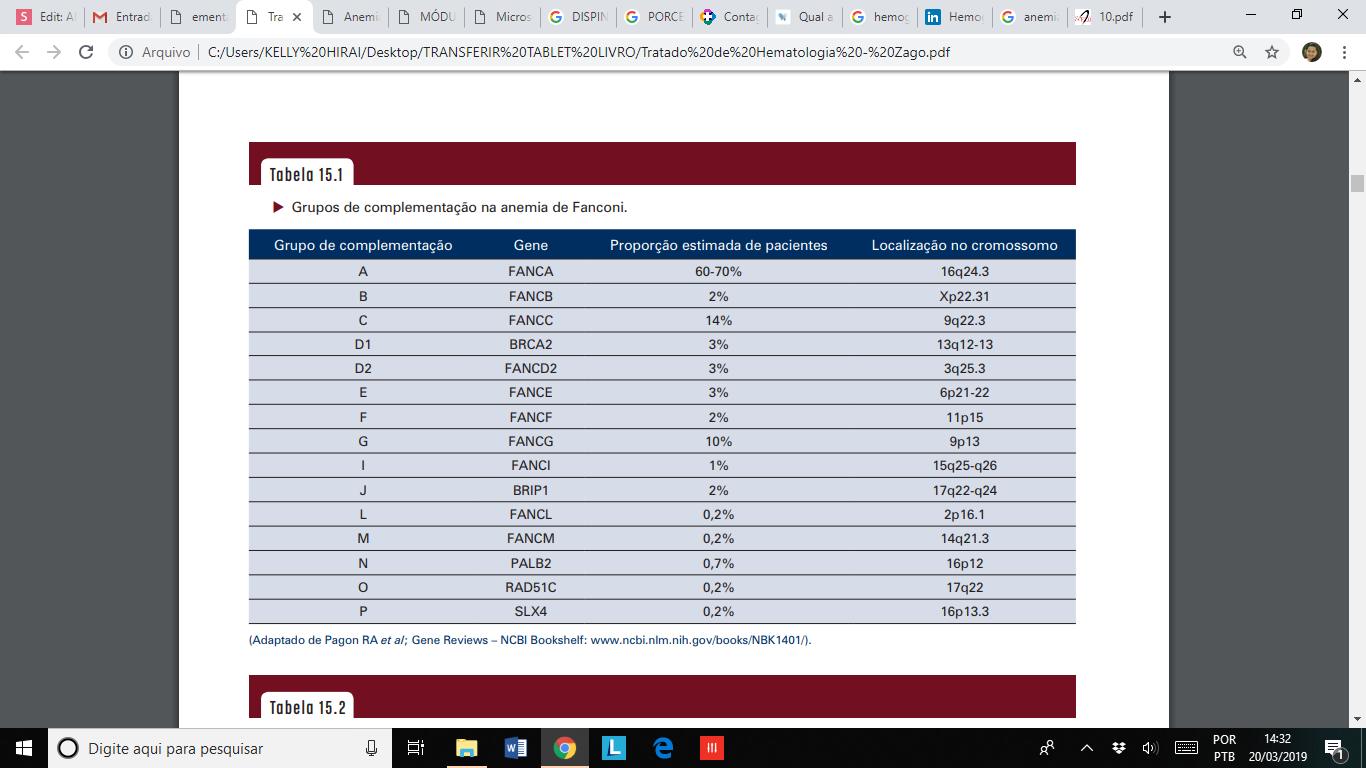
As mutações que ocorrem nos genes dos pacientes com AF impedem que o reparo ao DNA seja feito de maneira adequada e isso ativa a apoptose celular, leva a uma depleção das células-tronco hematopoéticas e causa pancitopenia.
Essa mesma dificuldade em reparar o DNA pode levar a mutações e translocações que resultam em mielodisplasia e/ou leucemia mieloide aguda.
As manifestações hematológicas aparecem ao redor dos oito anos de idade, e geralmente levam ao diagnóstico da doença, apesar de as malformações estarem presentes desde o nascimento.
As más-formações mais frequentes desta síndrome são: deformidades ósseas (ausência de polegar, polegar menor ou pendurado e ainda ausência do osso do braço chamado rádio - deixando o braço curto e curvo), o rosto delicado com boca pequena, olhos pequenos e queixo pequeno. A baixa estatura é frequente e geralmente as crianças nascem com baixo peso e estatura abaixo do esperado para idade. Os pacientes com frequência apresentam manchas na pele de cor café-com-leite.
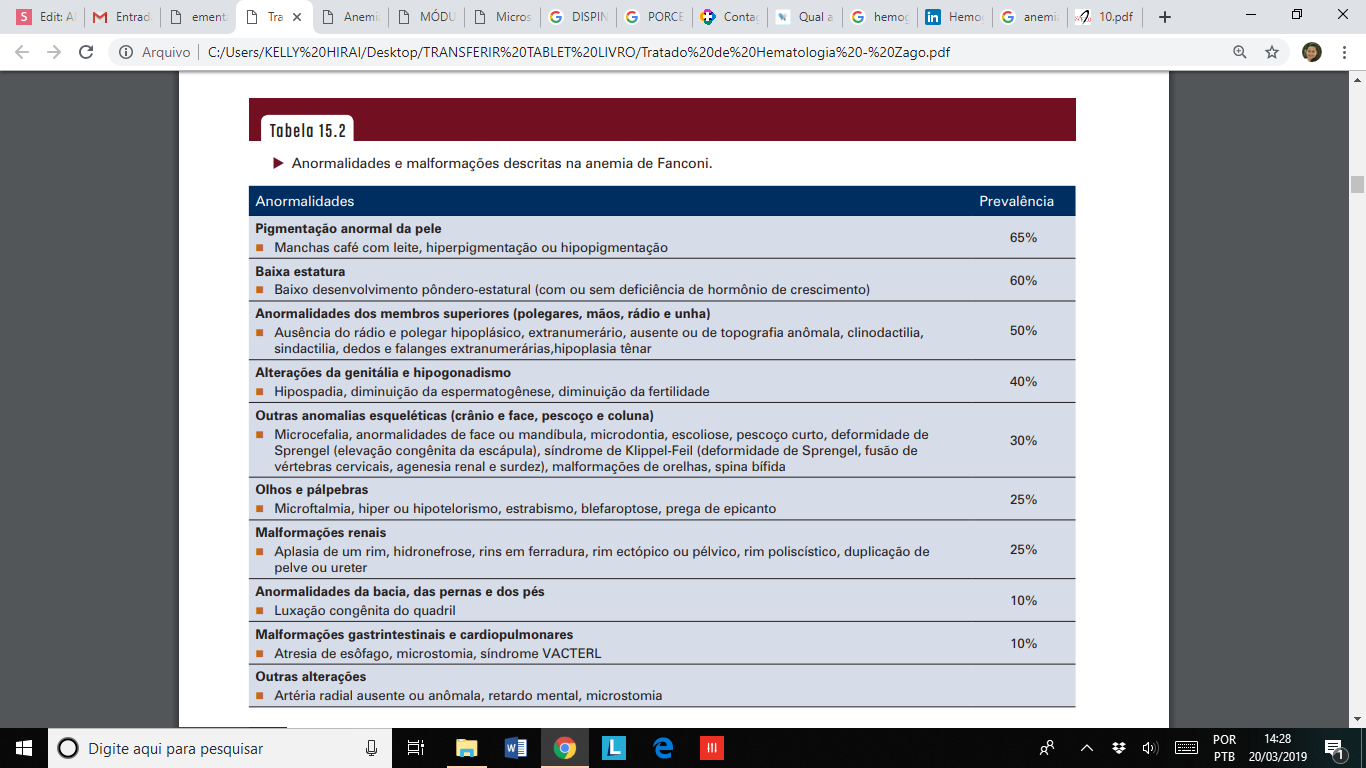
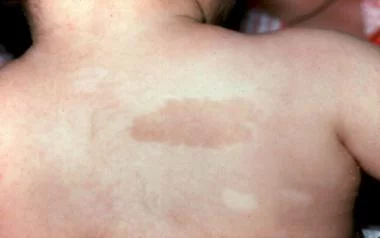

ASPECTOS LABORATORIAIS
Inicialmente os pacientes apresentam macrocitose e trombocitopenia e, a seguir, progridem para pancitopenia grave, configurando um quadro hematológico indistinguível da anemia aplástica grave. Raramente, alguns pacientes podem apresentar melhora espontânea dos sintomas hematológicos. A eritropoese é do tipo fetal (com aumento da hemoglobina F), e está associada a altos níveis de eritropoetina.
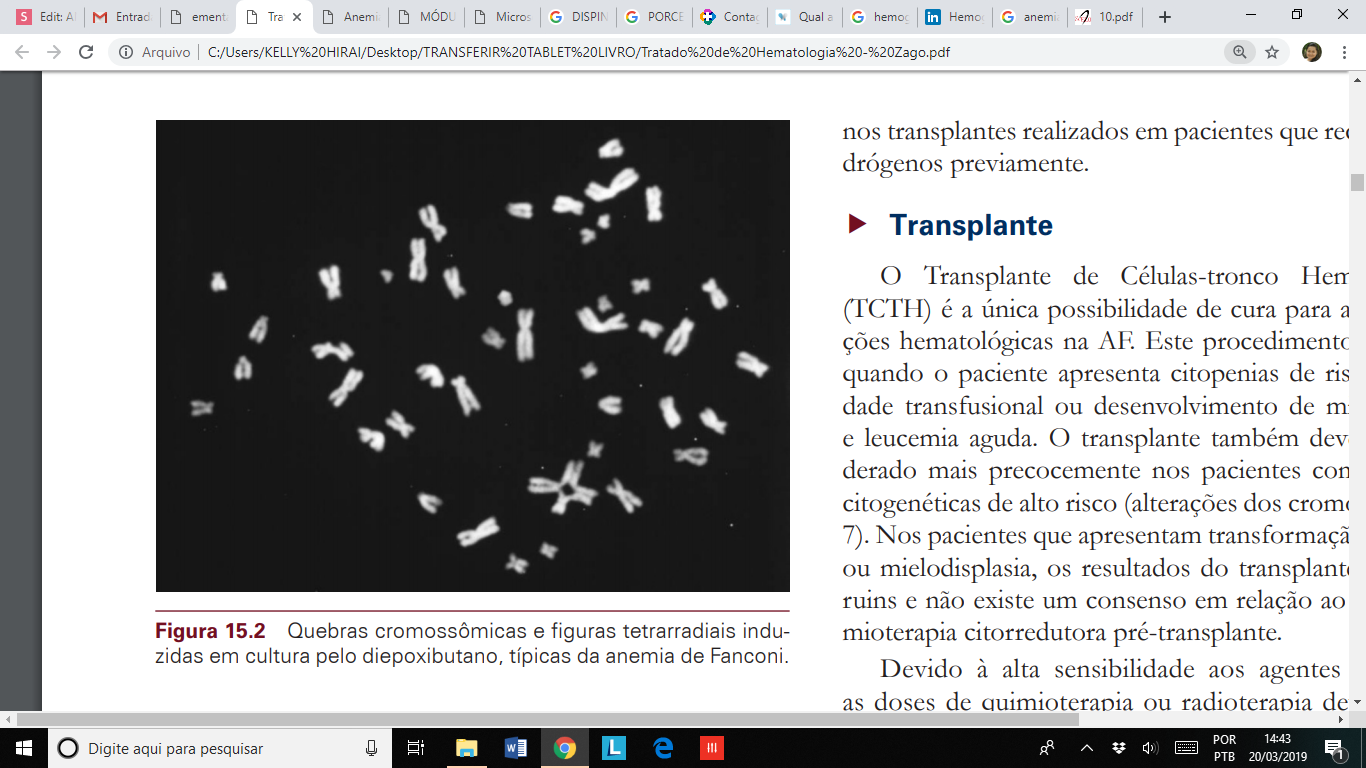
O teste que utiliza o diepoxibutano (teste do DEB) é altamente sensível e específico para AF.
Quando expostas a este agente, as células dos pacientes com AF apresentam inúmeras anormalidades cromossômicas como as endorreduplicações, rearranjos, falhas e quebras de isocromátides e de cromátides, anéis dicêntricos e figuras radiais entre cromossomos heterólogos tri, tetra e pentarradiais
HEMATOLOGIA CLÍNICA - ANEMIAS DECORRENTES DE DOENÇAS DA MEDULA ÓSSEA
By KELLY EMI HIRAI



