Teaching Demonstration
University of Pittsburgh
Feb 5, 2025
Alex Maldonado
BLAST

Typically, I have an anonymous TopHat discussion open while I teach to . . .
Encourage more questions
Gauge understanding
BLAST in the context of BIOSC 1540
The first half of the course would cover bioinformatics, and the second half would be simulation and modelling
BLAST
Content focuses on getting a brief introduction to the "why" and the "how" to prepare for future computational biology courses
1. DNA sequencing
2. Genome assembly
3. Gene prediction
4. Sequence alignment
5. Read mapping
6. RNA quantification
Python and FASTQ
Foundations (Tues)
Methodology (Thurs)
Greedy algorithm; De Bruijn graphs
Hidden Markov Models
Global and local alignments; FM-index
Seed-Chain-Extend algorithm
Generative models
...
...
Bite-sized assignments scaffold into a practical genomics project

Other projects include:
- Gene expression changes under microgravity (NASA)
- Protein structure prediction of DHFR
- Web-based docking for novel antibiotics
Students would already be familiar with the following concepts

How homology provides insight into gene function, evolution, and protein structure
Query 1 ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAA 60
Sbjct 2555705 .....A.....A..AA.T..C..T..C..TAAA...A....C.....G.ACC........ 2555646
Query 61 TTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACT 120
Sbjct 2555645 ...........CT.............A......A.....C..C.GA.C.....GA....A 2555586
Query 121 TTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAAT 180
Sbjct 2555585 C.T.......CA..G..A..T...A.T..T..A..G..G...T.G..A...A.A..T..C 2555526
Query 181 GTTGTACTTACTTC---AGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCT 237
Sbjct 2555525 ..C.....C...AACCA..C.T..--.....C.A.GA....-..A.....T..AA.C... 2555469
How to interpret alignments
Dynamic programming and pairwise alignment algorithms
H(i,j) = \max \left\{ \begin{array}{l}
0, \\
H(i-1,j-1) + s(x_i, y_j), \\
\max_{k\geq1}\{H(i-k,j) - W_k\}, \\
\max_{l\geq1}\{H(i,j-l) - W_l\}
\end{array} \right.
Students are familiar with these concepts
After today, you should have a better understanding of
Insights and limitations of pairwise sequence alignment
At this stage in our project, we have assembled contigs and predicted hypothetical proteins
Genome assembly
> hypothetical protein
ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAATTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACTTTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAATGTTGTACTTACTTCAGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCTATTGAAGATATTTATCAACTACCGGGCCATGTTTTTATATTTGGAGGGCAAACATTATTTGAAGAAATGATTGATAAAGTGGACGACATGTATATTACTGTTATTGAAGGTAAATTTCGTGGTGATACGTTCTTTCCACCTTATACATTTGAAGACTGGGAAGTTGCCTCTTCAGTTGAAGGTAAACTAGATGAGAAAAATACAATTCCACATACCTTTCTACATTTAATTCGTAAAAAATAAOpen reading frames
How could we determine the protein function this gene encodes for?
Staphylococcus aureus
Example coding sequence identified by prodigal
Paired-ended Illumina reads assembled with SPAdes
If our query sequence has high similarity to a known annotated gene, we assume similar functionality
Query 1 ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAA 60
Sbjct 1555151 ............................................................ 1555092
Query 61 TTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACT 120
Sbjct 1555091 ............................................................ 1555032
Query 121 TTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAAT 180
Sbjct 1555031 ............................................................ 1554972
Query 181 GTTGTACTTACTTCAGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCTATT 240
Sbjct 1554971 .....................................A...................... 1554912
Query 241 GAAGATATTTATCAACTACCGGGCCATGTTTTTATATTTGGAGGGCAAACATTATTTGAA 300
Sbjct 1554911 ............................................................ 1554852
Query 301 GAAATGATTGATAAAGTGGACGACATGTATATTACTGTTATTGAAGGTAAATTTCGTGGT 360
Sbjct 1554851 ............................................................ 1554792
Query 361 GATACGTTCTTTCCACCTTATACATTTGAAGACTGGGAAGTTGCCTCTTCAGTTGAAGGT 420
Sbjct 1554791 ............................................................ 1554732
Query 421 AAACTAGATGAGAAAAATACAATTCCACATACCTTTCTACATTTAATTCGTAAAAAATAA 480
Sbjct 1554731 ............................................................ 1554672Our hypothetical gene has a high similarity to dihydrofolate reductase from Staphylococcus aureus
Pairwise alignments identify optimal placement of matches, mismatches, and gaps
Review question
Query 1 ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAA 60
Sbjct 2555705 .....A.....A..AA.T..C..T..C..TAAA...A....C.....G.ACC........ 2555646
Query 61 TTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACT 120
Sbjct 2555645 ...........CT.............A......A.....C..C.GA.C.....GA....A 2555586
Query 121 TTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAAT 180
Sbjct 2555585 C.T.......CA..G..A..T...A.T..T..A..G..G...T.G..A...A.A..T..C 2555526
Query 181 GTTGTACTTACTTC---AGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCT 237
Sbjct 2555525 ..C.....C...AACCA..C.T..--.....C.A.GA....-..A.....T..AA.C... 2555469
Query 238 ATTGAAGATATTTATCAACTACCGGGCCATGTTTTTATATTTGGAGGGCAAACATTATTT 297
Sbjct 2555468 C....T..A...A.AG.GT..T.T..T....................A.....G....AC 2555409
Query 298 GAAGAAATGATTGATAAAGTGGACGACATGTATATTACTGTTATTGAAGGTAAATTTCGT 357
Sbjct 2555408 ....C.........CC.G..A..T..T........C..A..A..A..T..A..G....AA 2555349
Query 358 GGTGATACGTTCTTTCCACCTTATACATTTGAAGACTGGGAAGTTGCCTCTTCAGTTGAA 417
Sbjct 2555348 ..A..C..A...........A..C.....C...A..........C.AA........A... 2555289
Query 418 GGTAAACTAGATGAGAAAAATACAATTCCACATACCTTTCTACATTTAATTCGTAAAAAA 477
Sbjct 2555288 ...C..........A........T..A..G.....A..CT........G.G....G.... 2555229Which pairwise algorithm is used to find the optimal alignment across the entire length of two sequences?
A. Needleman-Wunsch
B. Smith-Waterman
C. De Bruijn
D. Clustal Omega

| D | ||||||
|---|---|---|---|---|---|---|
1
0
2
3
4
5
0
1
2
3
4
5
0
0
A
T
T
A
C
A
A
T
T
C
0
0
0
0
0
0
0
0
0
1
1
0
0
0
0
2
1
0
0
0
1
3
2
0
1
0
2
2
1
0
0
1
3
0
These algorithms guarantee optimal local alignment for sequence comparison
Students are familiar with these concepts
Dynamic programming is used to fill the scoring matrix
Example: Smith-Waterman
and tracebacks provide the highest-scoring alignment(s)
A
T
T
A
T
T
A
T
T
A
T
T
A
C
-
C
Applying pairwise alignment to massive databases is computationally infeasible
1 5 10 15 20 25
1 AGATGATAGTCGCAAGCCTTGACCTCATAT
2 GCGGATGTCAATGAGTTTGCGTGCAGCTAA
3 TTTTAGTTGCAGTTTGTGATGTCCTGCAGT
4 ACTAGCTACACATGCAGTTCTGATTTGCGT
5 AAACTATTAGAATCTTCCAGTTCGTACCTG
6 GCCCGATAAGCTTGCCGAGCACTGGTGCCG
7 TTTGGAGCGCCGTACCTGGTGAGCTAACGG
8 CTTACCCCCGATTCGCGACCGTAATTGCAT
9 TTGCAAAGCTACCCGTAGCGGACATTATAG
10 ACATTCCCATCAGCTAAATCTTGCAGGTCG
11 TTTGCAAGCTAACGTATATACCAATGATAG
12 TCAAGCTACGTTACTTGGAGGCATTGCAGC
13 AGTAGTTATTACCTGAAGCACATTACAGGA
14 CAGCTAGTTTCTTAATCTATTGCTAGAACA
15 ATATCGCAACAATAGCTAACCCTTTCACTCAACAGCTAATGCCGCTTGCAATC
For example:
AGCTA
...
TTGCA
We are searching for a sequence that contains conserved regions of
in this exact order
Rows 10 and 12 are possible hits
Let's do a word search!
Systematic searches take a long time!
Ready?
Go!
Note: There are two possible rows
For about 30 seconds
After today, you should have a better understanding of
Speed and capabilities of BLAST
BLAST prioritizes speed over exact optimality, making large-scale searches feasible
Basic Local Alignment Search Tool (BLAST) uses heuristics to identify high-probability matches
BLAST does not guarantee optimal alignments like in Smith-Waterman, but it is orders of magnitude faster

Let's see an example
> hypothetical protein
ATGACTTTATCCATTCTAGTTGCACATGACTTGCAACGAGTAATTGGTTTTGAAAATCAATTACCTTGGCATCTACCAAATGATTTGAAGCATGTTAAAAAATTATCAACTGGTCATACTTTAGTAATGGGTCGTAAGACATTTGAATCGATTGGTAAACCACTACCGAATCGTCGAAATGTTGTACTTACTTCAGATACAAGTTTCAACGTAGAGGGCGTTGATGTAATTCATTCTATTGAAGATATTTATCAACTACCGGGCCATGTTTTTATATTTGGAGGGCAAACATTATTTGAAGAAATGATTGATAAAGTGGACGACATGTATATTACTGTTATTGAAGGTAAATTTCGTGGTGATACGTTCTTTCCACCTTATACATTTGAAGACTGGGAAGTTGCCTCTTCAGTTGAAGGTAAACTAGATGAGAAAAATACAATTCCACATACCTTTCTACATTTAATTCGTAAAAAATAABLAST dramatically reduces the computational cost of sequence querying
NCBI has more than 254 million DNA sequences as of December 2024
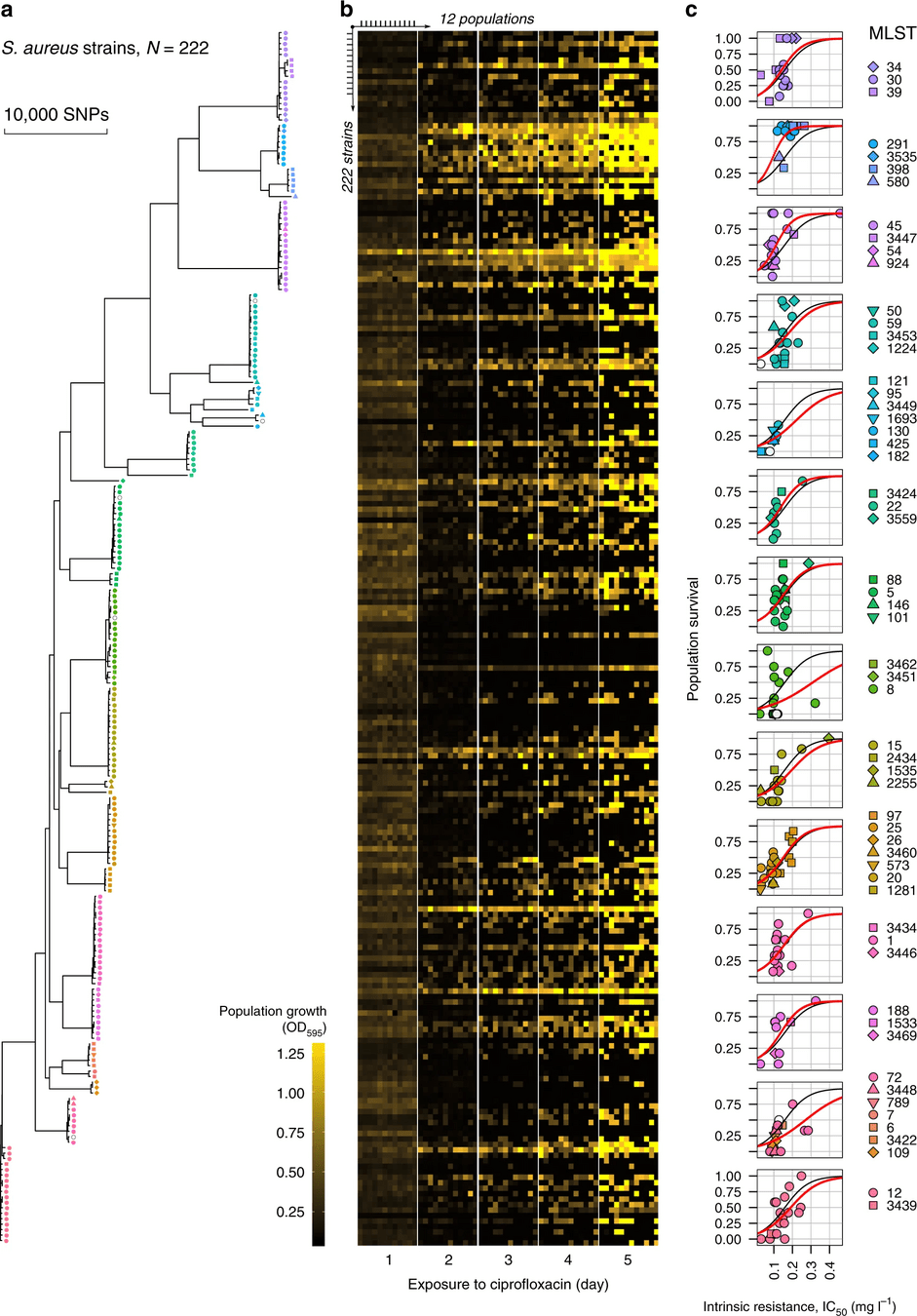
This enables all kinds of research, such as our project to elucidate how S. aureus was evolving ciprofloxacin resistance
After today, you should have a better understanding of
Seed-and-extend algorithm
Pairwise alignment is similar to computing the similarity of every sentence on every page
Suppose I ask you to search for the sentence, "... intron and exon (acceptor splice site)" in a textbook


How would you do this?
We need a method that avoids checking every sentence individually
We could use the index to find relevant pages to find individual words!

What could we use to help you find this sentence faster?

In the context of BLAST, we create a lookup table to identify likely sequences to align
Lookup tables provide a place to start searching from
1 5 10 15 20 25
1 AGGCTTGCATCCCACCAGCTAAAATTGAAT
2 TGGTACGCAATGTTTGAGGGAGTCGCATTC
3 AACAGCTATCTTTTGAAGAGTCCTTGCATC
4 GCTACAAAAACTTCATTTGCATACACGGAA
5 TGGGTTACCGCTAGCCATGGAATTAAAAGC
6 CCCAGTTTCATCCTTTGAAGCTTGCATGCT
7 GAGGTTTTAGGCTAGAGGGTGTTGGACAAT
8 CGTTTAGCTACCGGGGCATGCGGGCTACGG
9 TGGAGTGTATTCATTGCGAGCTAGCTTGGA
10 ACTTACGTAGCAGAGTAATAGCTATTATAC
11 AGATGCAACTAAACATACTCATTCTTCAAA
12 GTTGCAGCTCGGGAGAGGTGATTAGTGTGA
13 CCAGTCATGCTAACAAGCAACTGTGTTCAG
14 AGACATATTGGAGCTAACTGAGCTGTTGCA
15 CGTATTGCTTTGCAGTTATAATCCGCTCGCHowever, now we will use a lookup table where I will tell you the rows these k-mers are located
Rows 3 and 14 are possible hits!
Let's do another word search!
We are searching for the same conserved sequence
AGCTA
...
TTGCA
AGCTA:
This is much faster!
Ready?
Go!
TTGCA:
1, 3, 8, 9, 10, 14
1, 3, 4, 6, 12, 14, 15
For about 30 seconds
Note: There are two possible rows
BLAST uses k-mer word matching to quickly narrow down the search space
Step 3: Efficiently scan NCBI databases to check if there is a matching k-mer in our query sequence
Step 1: Query is broken into overlapping k-mers.
Step 2: Take each k-mer and build a hash map (i.e., dictionary) containing locations within the query sequence
| k-mer | Index |
|---|---|
| AAC | [3] |
| TAA | [2] |
| GCT | [0, 5] |
| ... | ... |
Students are familiar with these concepts
Requires highly optimized C++ code that is beyond the scope of this course
GCTAAGCT
k = 3
GCT
CTA
TAA
AAG
AGC
GCT
Lookup is O(q) with O(M x d) scanning
After identifying matching k-mers, why should we even align the sequences?
Which algorithm should we use?
Talk with your neighbors!
After today, you should have a better understanding of
Seeding the lookup table
Commentary: This would start covering our "methodology" lecture
from collections import defaultdict
def build_lookup(sequence, word_size):
lookup = defaultdict(list)
for i in range(len(sequence) - word_size + 1):
kmer = sequence[i:i+word_size]
lookup[kmer].append(i)
return dict(lookup)
# Example usage:
query = "ATGCGTACGTTAGC"
lookup_table = build_lookup(query, word_size=5)BLAST first breaks the query sequence into words (k-mers) to create the hash map
Students are familiar with these concepts
1. Initialize a dictionary which defaults to an empty list
2. Loop through each k-mer in the sequence
2a. Add the index of the k-mer
3. Return our lookup table
Commentary: This is an option for teaching computational methods, but it is not as engaging
Instead, we explore the algorithms with Python in real time
Let's just walk through how I would lead this
Pitt Teaching Faculty Teaching Demo
By aalexmmaldonado




