Metagenome Program
Adam R. Rivers
JGI Scientific Advisory Meeting
January 19, 2016
Outline
-
Program overview
-
Program publications
-
Program science
- Viral discovery and machine learning
- Historical reconstruction with metagenomics
-
Improvements to user science
- Automating Stable Isotope Probing (SIP) metagenomics
- Community profiling with iTags
- metagenome assembly and binning
- Global metagenome comparisons
Interconnected
Function driven
Genome centric
Metagenome program overview

Metagenome program overview
- Data Products
-
Community iTags 10,000
-
Metagenomes 825
-
Metatranscriptomes 900
User driven science
Program driven science
- Metagenome program science
- Viral discovery and function
- Historical reconstruction
- Machine learning for metagenomics
- Microbial systems group science
- Plant microbe interactions
- Wetland biogeochemistry
Metagenome program in context



Carbon Cycling
Biofuels
Biogeochemistry
Integrated, system based approaches to understanding:
- Development of cross-platform analyses
- Integration of data across projects
- Research into carbon cycling and plant-microbe interaction
Metagenome program projects
Metagenome program publications
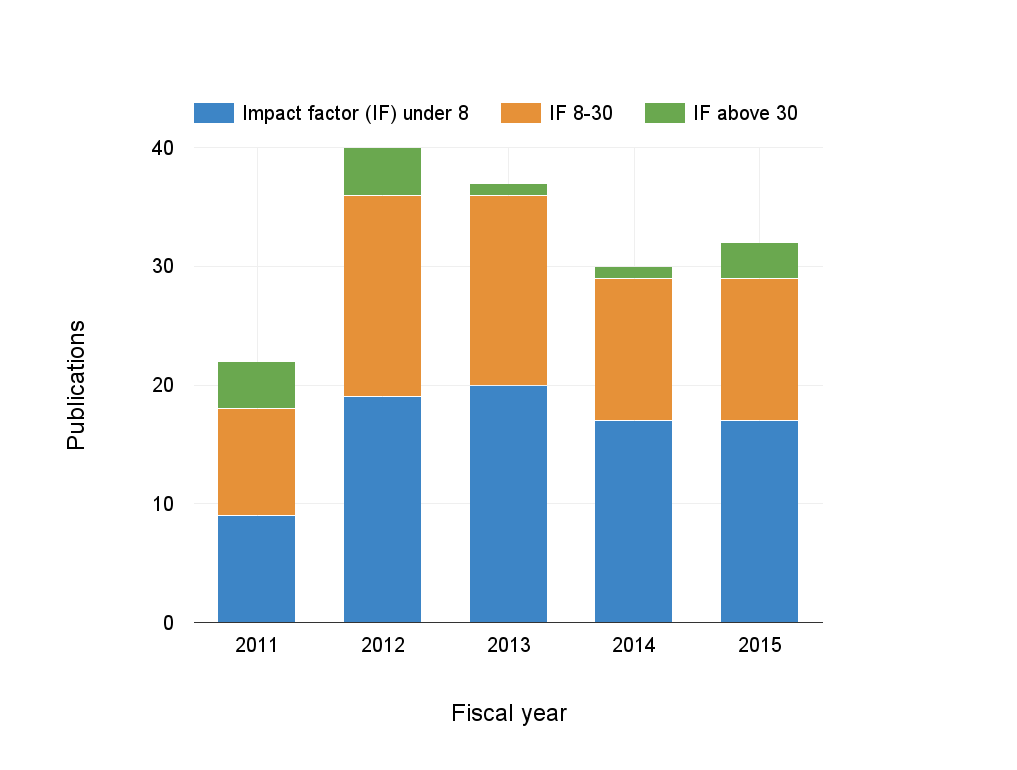
Metagenome program publications
Discovery of canidate radiation phyla
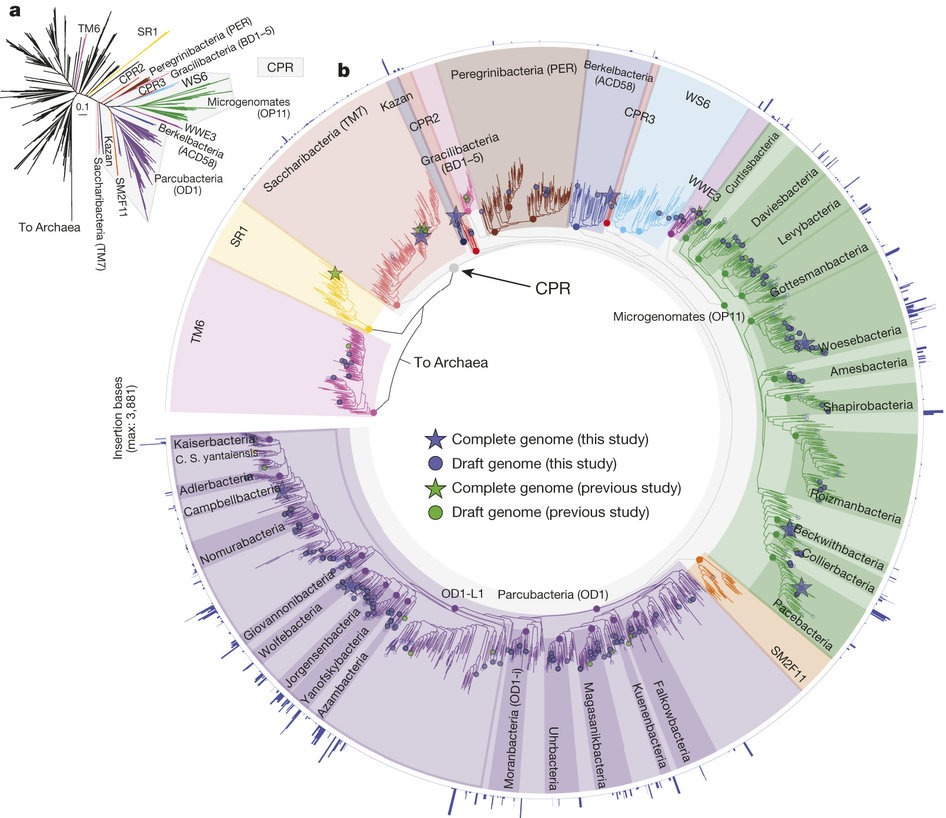


- A large group of uncultivated phyla are sound in groundwater
- Small cells, minimal genomes
- The phyla have highly divergent 16S genes with introns
Metagenome program publications
Salicylic acid modulates root colonization

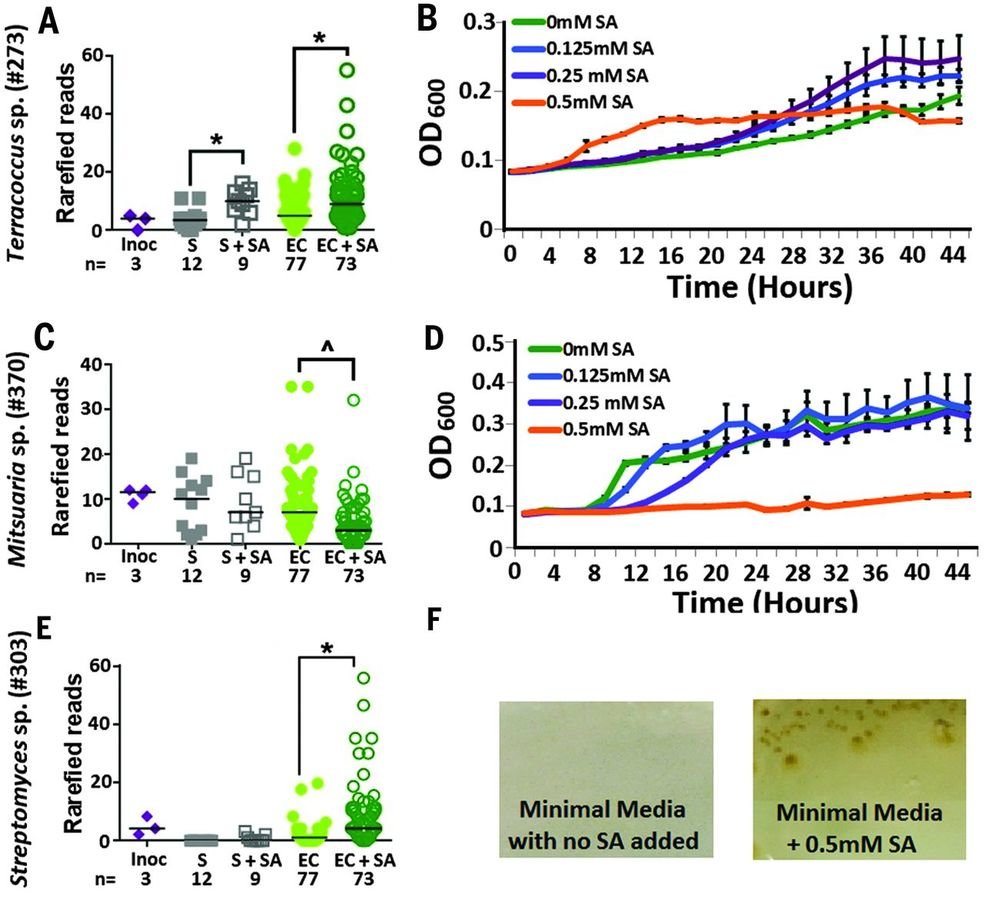

The discovery of a mechanism for allowing colonization by endophytic bacteria
Metagenome program publications


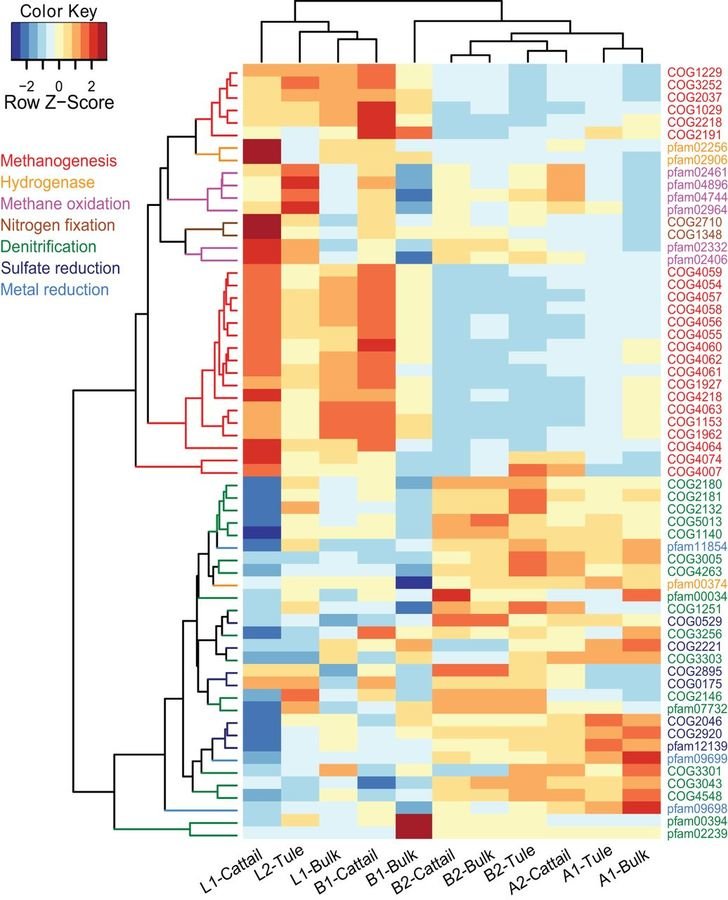
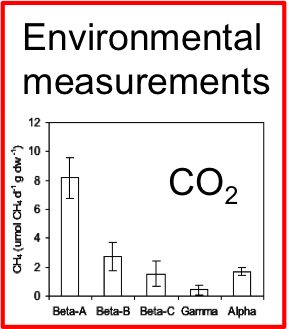
Methane production in restored wetlands
Does restoring wetlands help or hurt climate change?
Methanogen abundance and methane emissions from new wetlands are dependent on electron acceptors, salinity and age
Metagenome program science
Viruses as ecosystem drivers
Viral discovery in metagenomes and metatranscriptomes by machine learning
The first soil virus metagenome
The first "complete" virus metagenome (single and double stranded DNA and RNA viruses)
Diel infection of RNA viruses in lakes
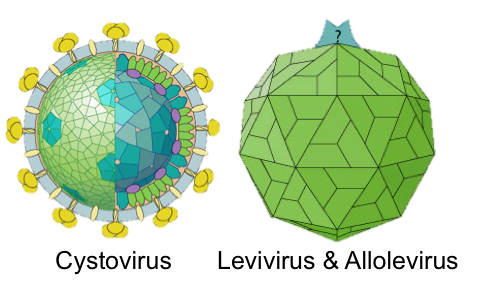


Finding highly divergent RNA viruses
Higher information content
Less bias

- Viral classifier with 95% recall
- Increasing precision by incorporating homology information to identify known organisms
- Screening of all metatranscriptomic data from IMG and Tara Oceans
Machine learning applications
- Supervised
- Supervised
- Unsupervised
- Unsupervised
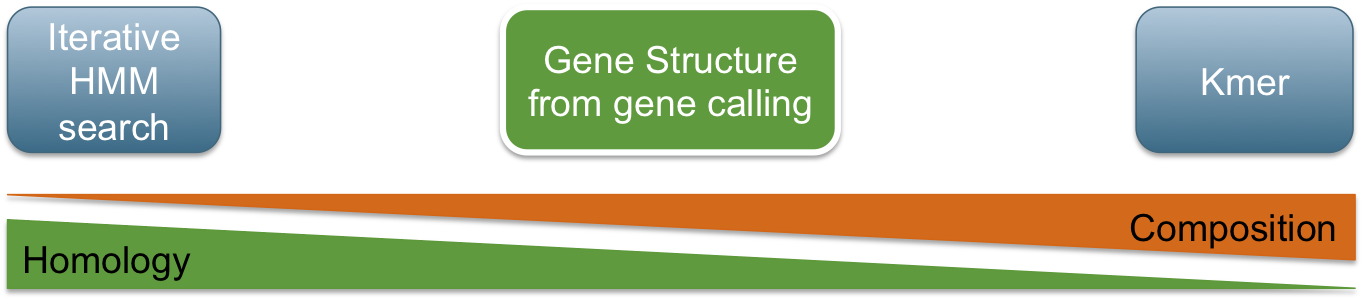
Classification in metagenomics
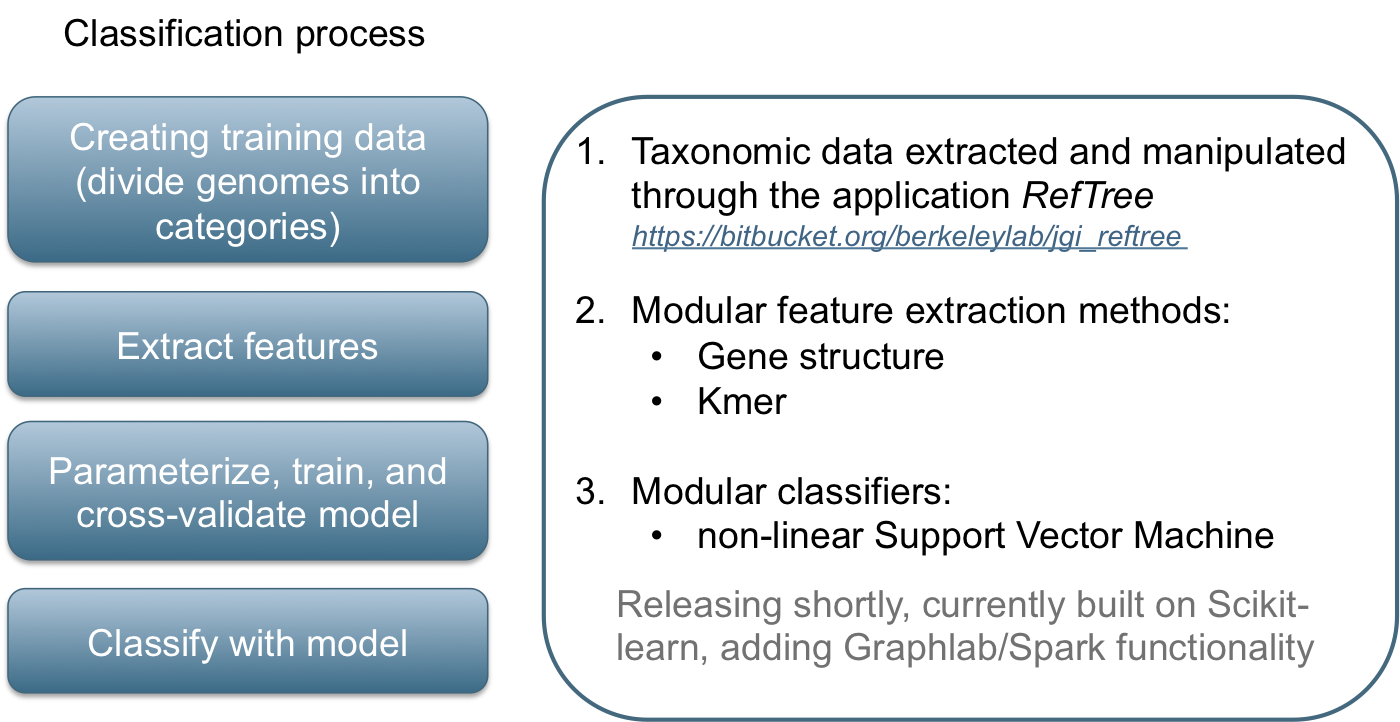
GeneLearn
A modular application for machine learning from sequence data
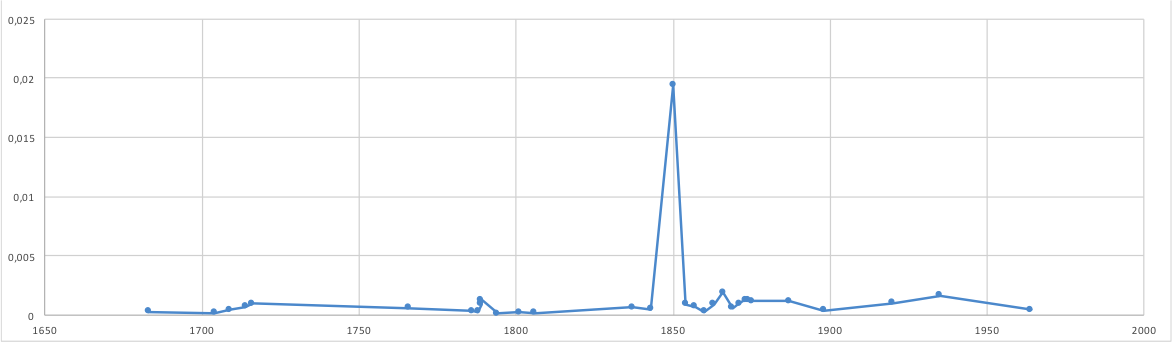
Historical reconstruction
metagenomics may have the ability to reconstruct past events leading to an understanding of for understanding climate, agricultural and human change
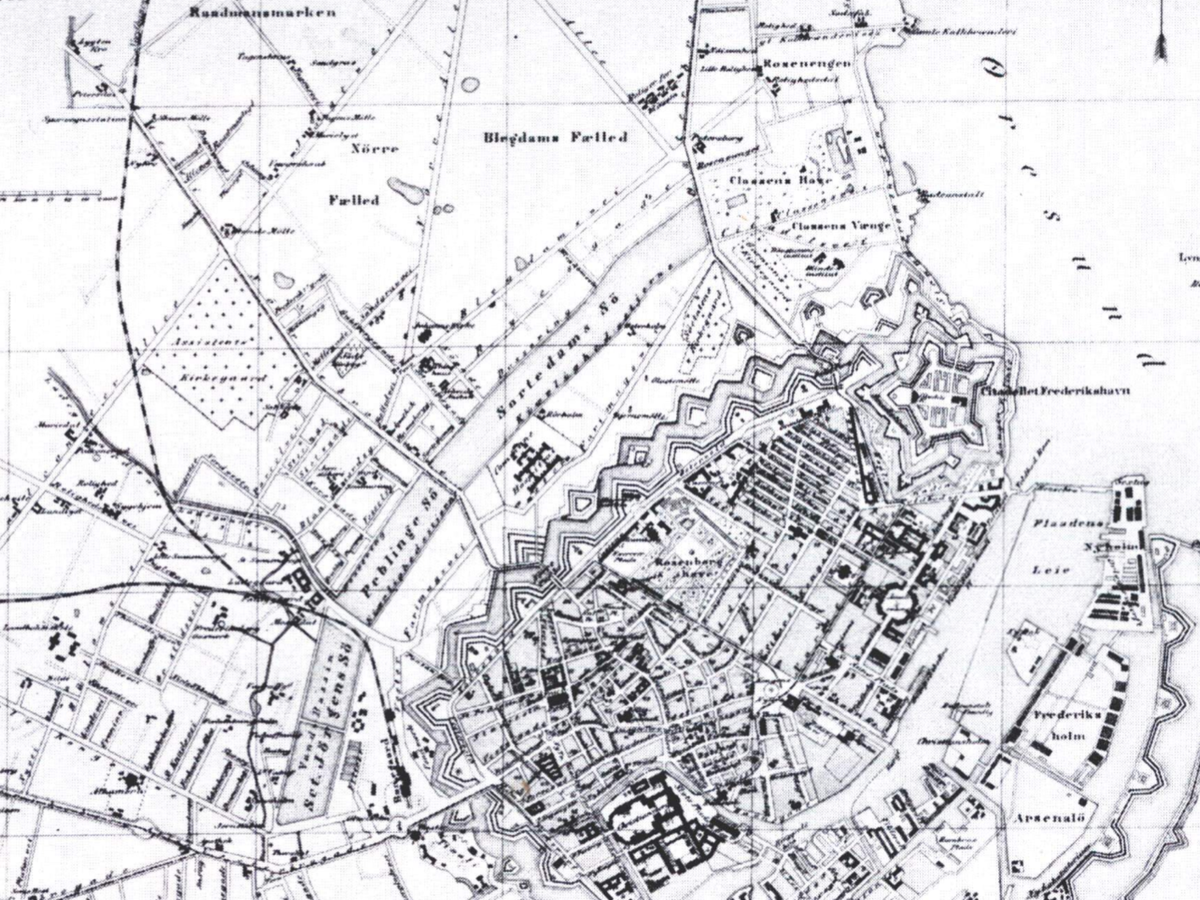
Historical reconstruction

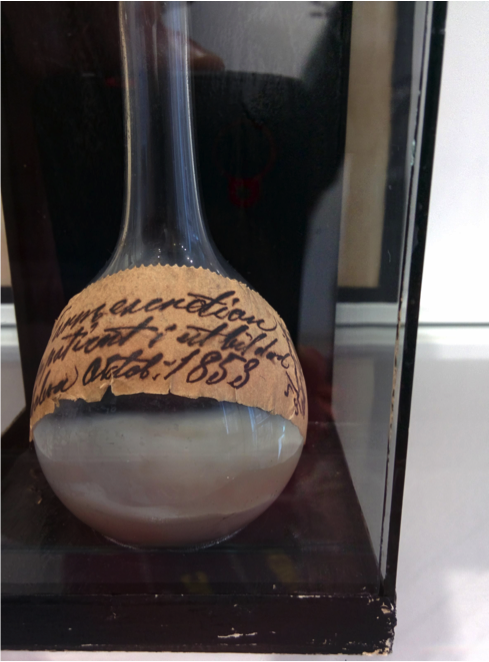
Construction 1649
British seige 1803
Cholera 1853
Cholera sequence
Improvements to user science
Interconnected
Genome centric
Function driven
iTags overhaul
Genome binning
Improved assembly
SIP ETOP
Host DNA depletion
Expression analysis
MT insert size
Gaia assembler
Interconnected
Function driven
Genome centric
Automating stable isotope probing

Stable isotope probing (SIP) is a method to identify the genes of microbes using a specific compound
SIP has been too complicated and time consuming to be widely adopted. This ETOP simplifies SIP to make it more widely available to JGI users


Jennifer Pett-Ridge
unlabeled DNA
labeled DNA
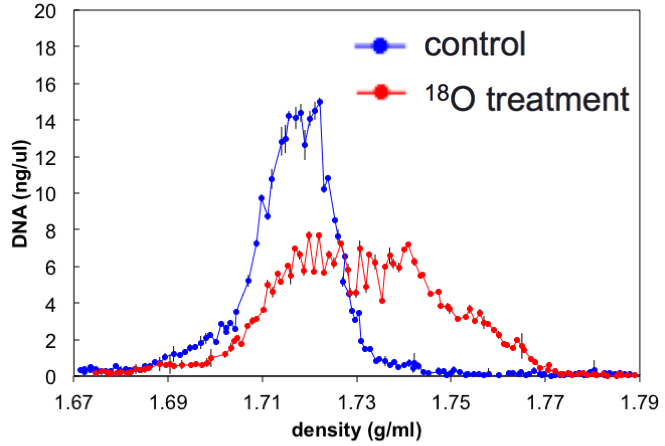
SIP automation ETOP
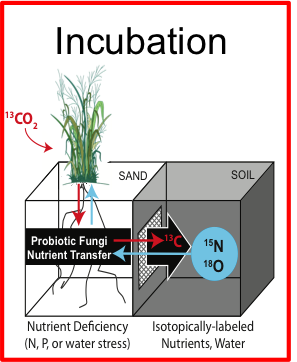
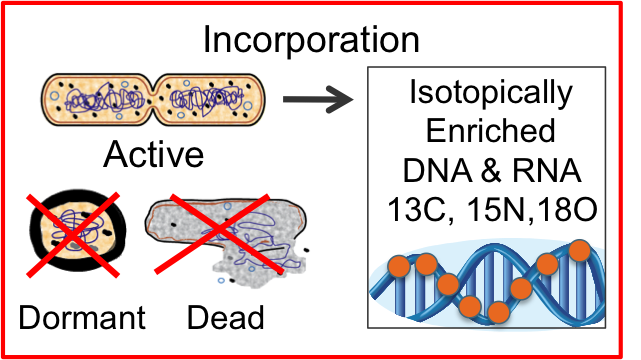

SIP automation ETOP

Current SIP-’omics approach is low-throughput
and requires special equipment
LLNL approach will use NanoSIMS for sample
prescreening prior to SIP processing/sequencing
Will also improve density gradient separation with
intercalators
And will automate:
-
Fraction collection
-
Density profile Characterization
-
Fraction cleanup (desalting)
-
Nucleic acid quantification
-
Reverse transcription and amplification
Total hands-on processing
| Standard SIP | ETOP SIP | |
|---|---|---|
| 1 sample | 13 | 1 |
| 24 sample experiment |
312 | 24 |
Itags amplicon sequencing
25,000 Itags sequenced since 2013
Itags are a useful, cost effective way of profiling communities but they are not being fully used.
Phase 1: Sequence the V4 and V5 region
Phase 2: Integration of all itag data and enhanced analysis
- Open Reference OTU picking across all JGI Itag data
- Improved metadata search and visualization tools
- More analyses e.g. Bayesian ecological networks
Metagenome assembly and binning
- Complete overhaul of Metagenome and Metatranscriptome assembly reduced resource use and increased assembly quality
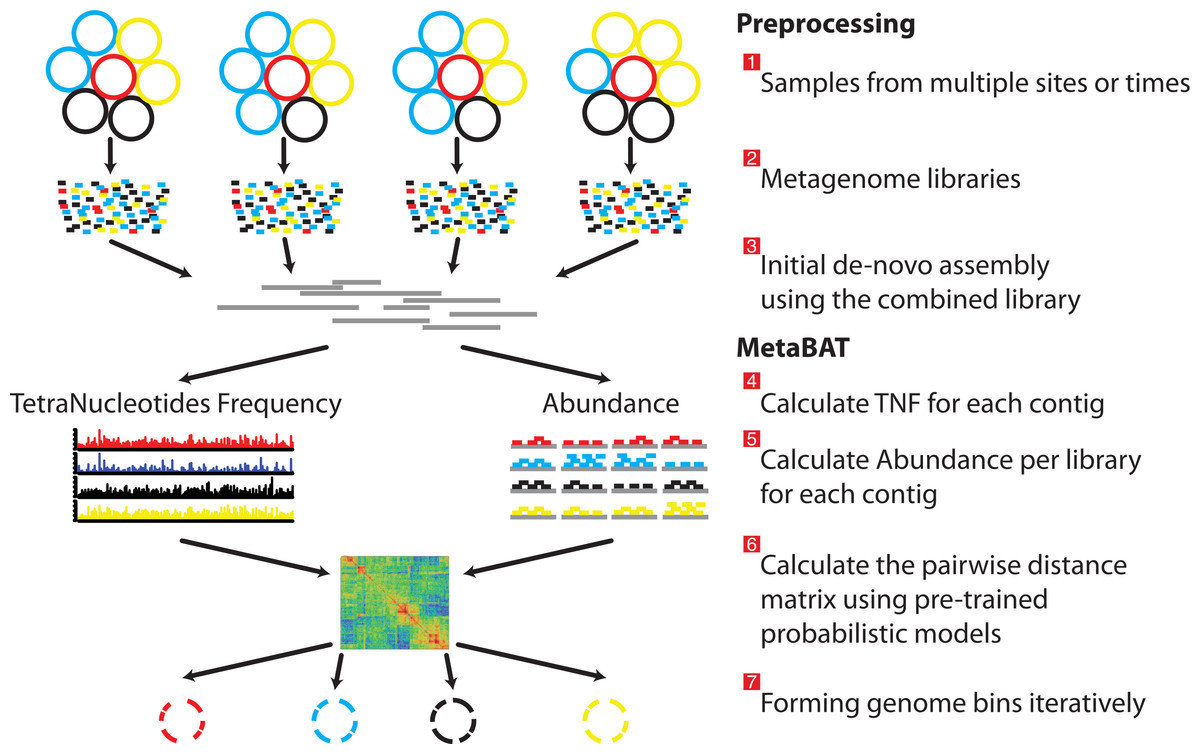
- Publication of Metabat for automatic genome binning
- Publication of Elviz for manual binning and exploration
All vs. all metagenomic assembly
- Metagenomic data are only compared against reference reads
- The cost of recomputing annotations when new references are added is high
The challenge:
The Gaia Assembler is a global, distributed, asynchronous assembly and alignment program designed to continuously align and annotate read data of arbitrarily large size.
Host Genome depletion
The challenge:
Endophytic bacteria cannot be effectively sequenced because host DNA overwhelms the sample

Working with a commercial partner to develop host specific depletion probes from genomic DNA
Charge Questions
Should the metagenome program prioritize functional methods like SIP and metatranscriptomics over genome reconstruction improvements?
Should we begin directing more resources towards developing methods to reconstruct metabolic networks?
Should more work be done to make metagenome data available in a machine readable way, even if that means that fewer interactive tools are available?
SAC presentation Jan. 20, 2016
By Adam Rivers

