Практическая работа с данными Hi-C
Александра Галицына
Цикл лекций о структуре хроматина
3 декабря 2020




Пространственная организация хроматина
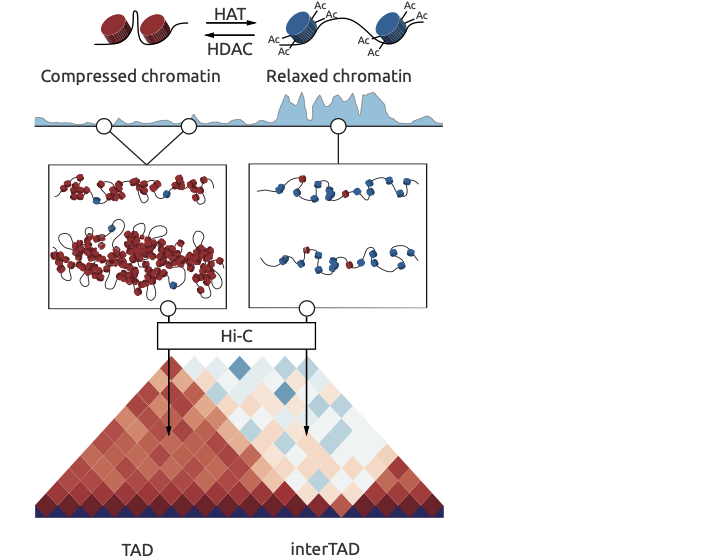
Ulianov et al. Genome Biology 2016

Подходы к изучению структуры хроматина
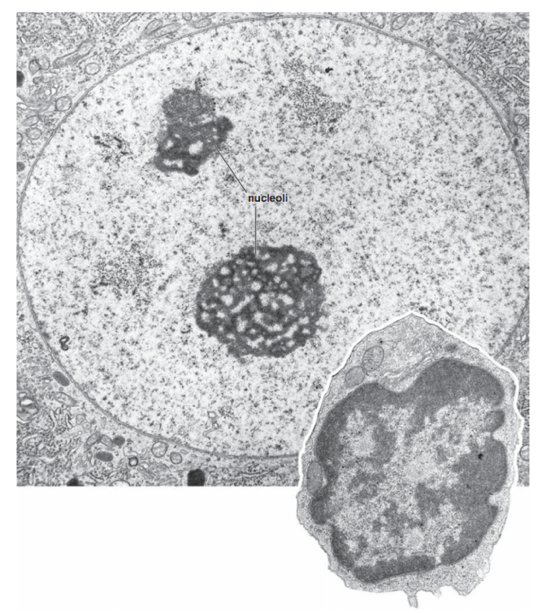
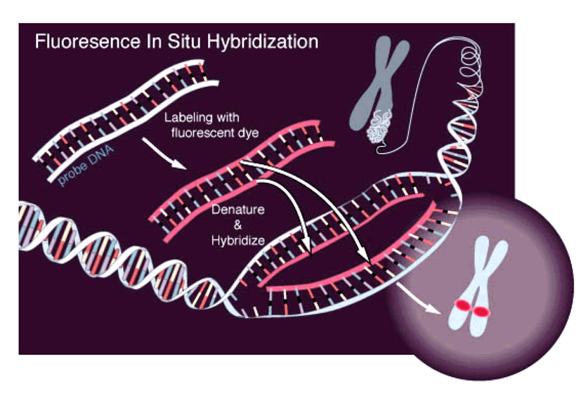
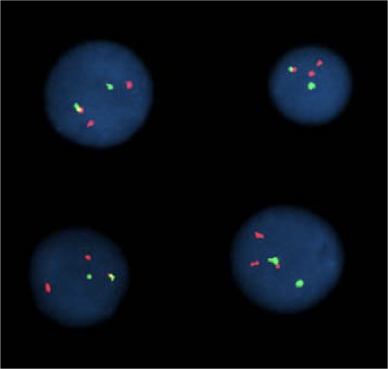
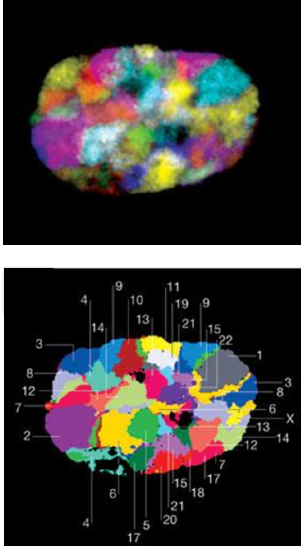
Микроскопия:
Микроскопия с флуоресцентными маркерами (FISH):
FISH на двух метках:
FISH для полных хромосом:
Chromosomes conformation capture (3C)
3C: Dekker et al. Science 2002
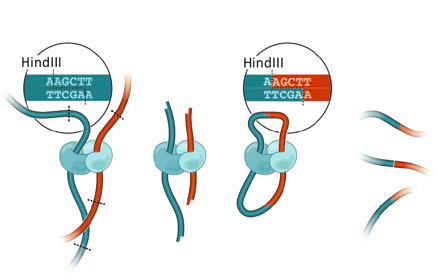
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Библиотека ДНК-ДНК контактов
Фиксация конформации хромосом
Hi-C (high-throughput CCC)
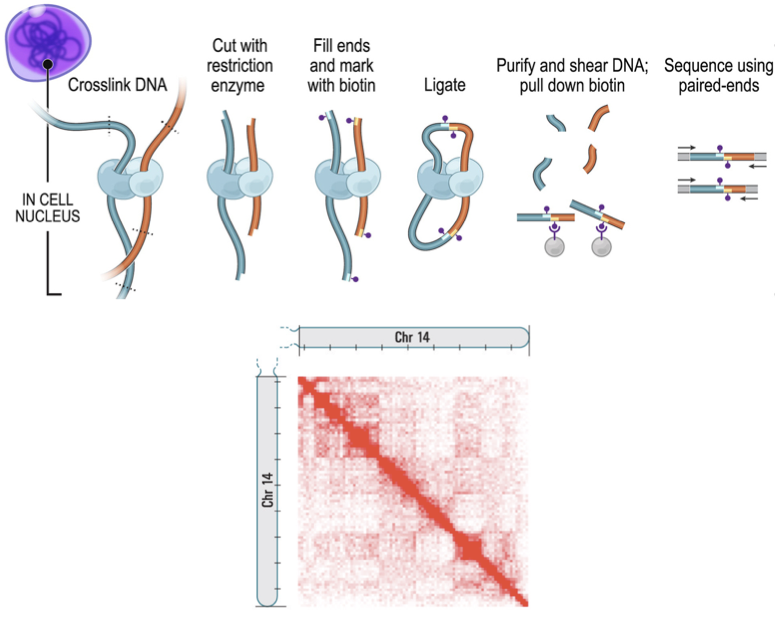
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Секвенирование
Картирование
Лигирование
ВЫСОКОпроизводительная фиксация конформации хромосом
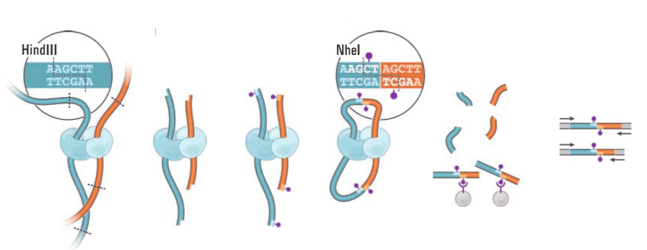
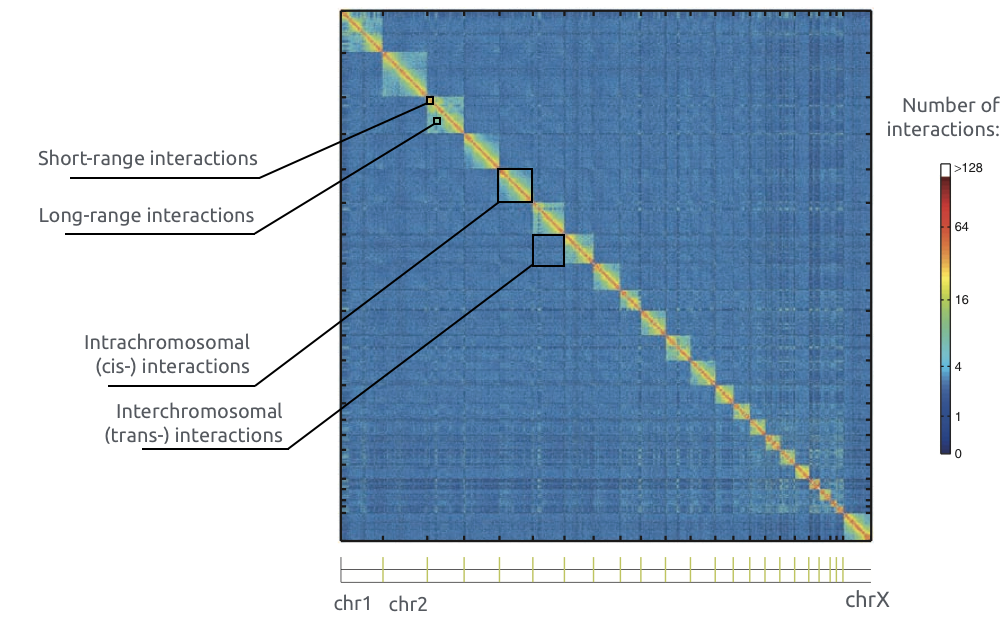
Разнообразие методов фиксации конформации
Adopted from Schmitt Nature Reviews 2016
Принципы организации структуры хроматина
Adopted from Imakaev et al. Nature Methods 2012
Общий план обработки Hi-C
0. Контроль качества данных NGS
1. Картирование ридов:
-> получение файла с картированиями (.sam)
2. Конвертация файла с картированиями (parse):
-> получение файла с парами контактов (.pairs)
- дедупликация
3. Получение карты геномных контактов (.cool):
- бинирование
- нормализация и корректировка
5. Контроль качества Hi-C
6. Поиск особенностей (фич) карт Hi-C
<- в следующий раз
Особенности данных Hi-C
! Секвенирование парноконцевое
Фиксация формальдегидом
Рестрикция ДНК
Лигирование
Очистка ДНК
Лигирование
Структура пары прочтений Hi-C

хороший сценарий картирования
риды (.fastq)
выравнивания (.sam/.bam)
контакты (.pairs)
тип контакта (техническая информация pairtools)
Структура пары прочтений Hi-C

не столь хорошие сценарии картирования...
картирование не произошло
Структура пары прочтений Hi-C
не столь хорошие сценарии картирования...
множественное картирование

Структура пары прочтений Hi-C
не столь хорошие сценарии картирования...
химерные прочтения - один рид перекрывает несколько фрагментов генома
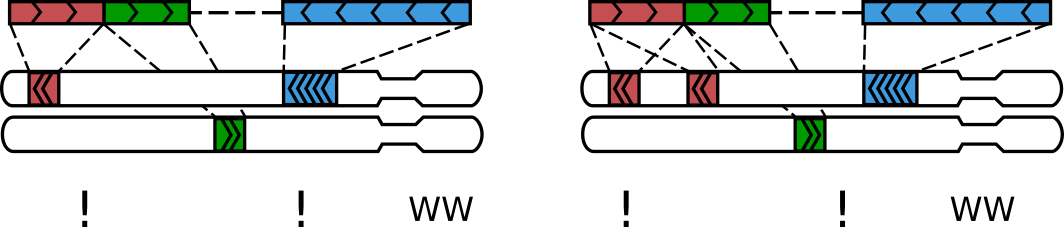
Структура пары прочтений Hi-C
Сквозные прочтения
(readthrough)
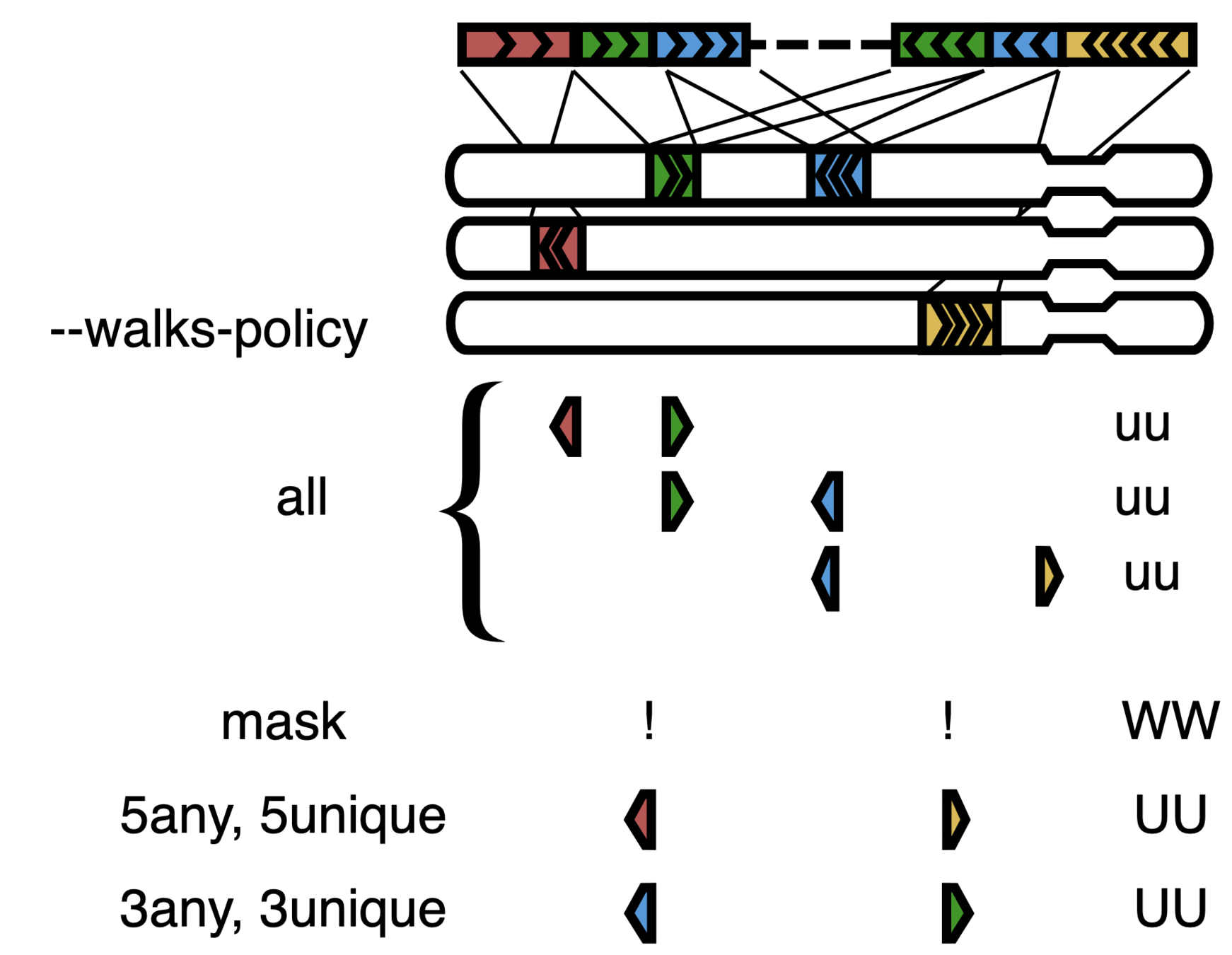
Получение карты контактов
Бины генома - это последовательные окна геномных координат одинакового размера.
Каждая ячейка (или пиксель) контактной карты содержит количество всех контактов, пришедших рестриктных фрагментов, соответствующих таким бинам:
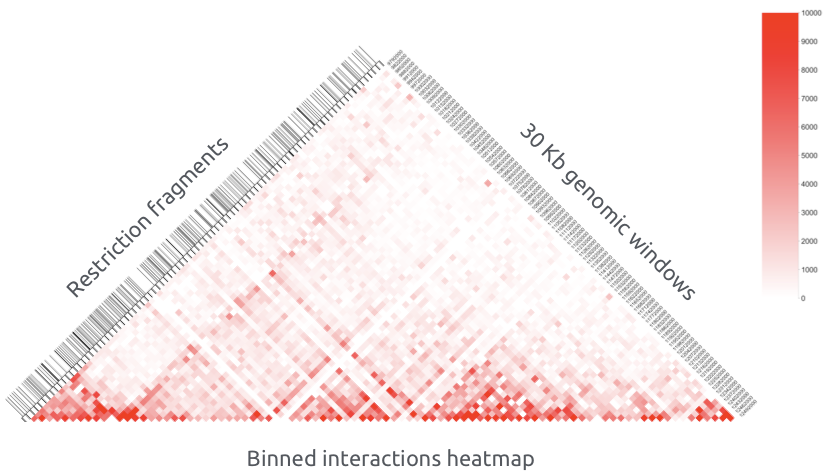
Бинированая карта контактов
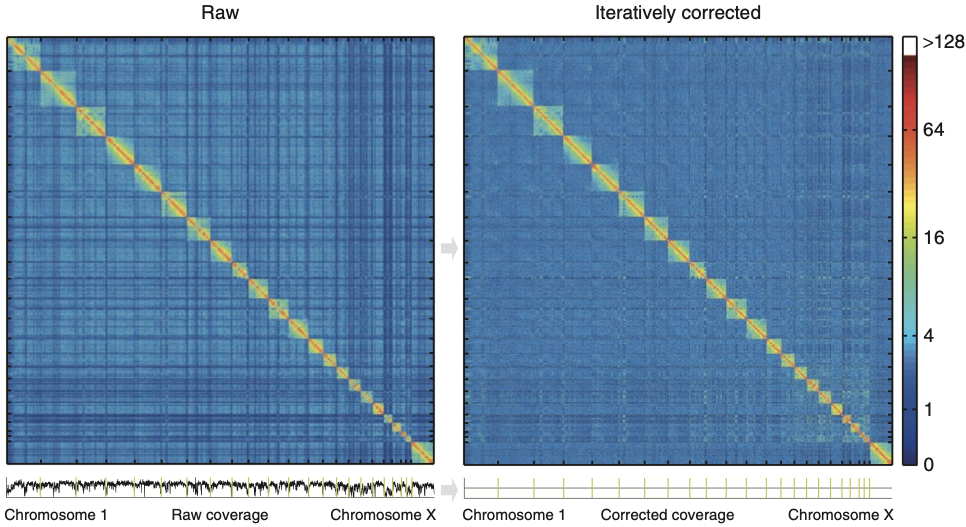
Нормировка и корректировка Hi-C
Imakaev et al. Nature Methods 2012
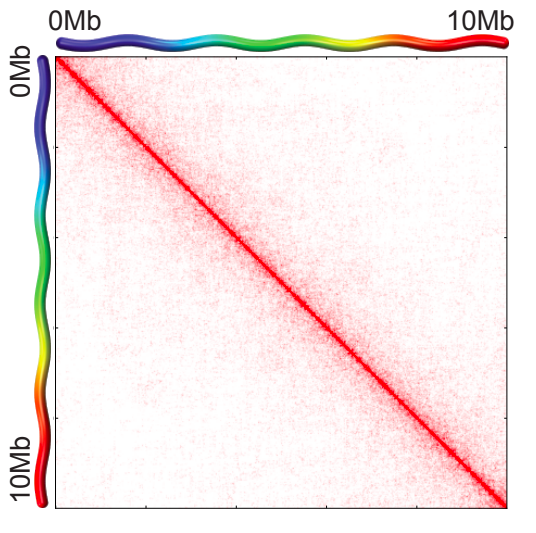
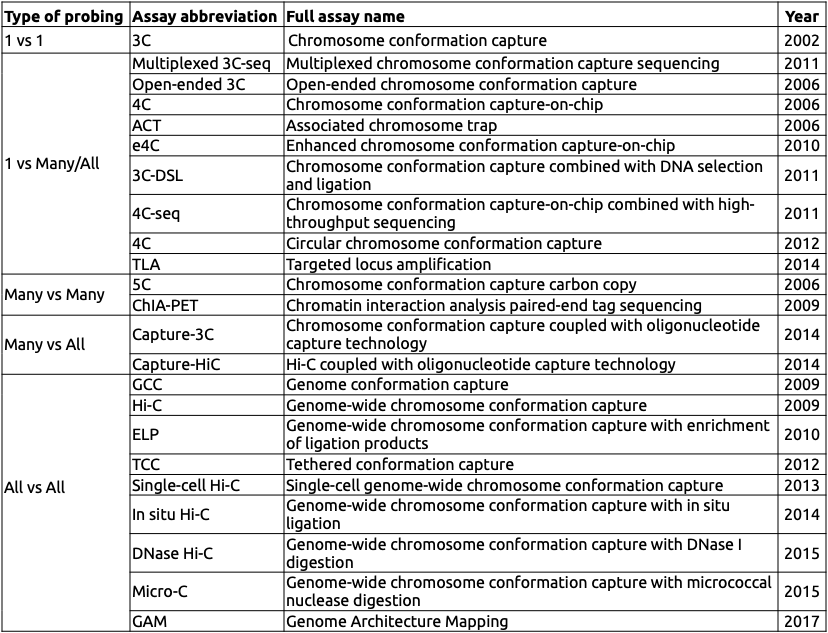
Графики P(s)
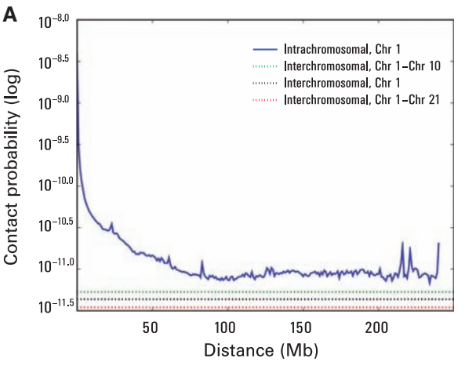
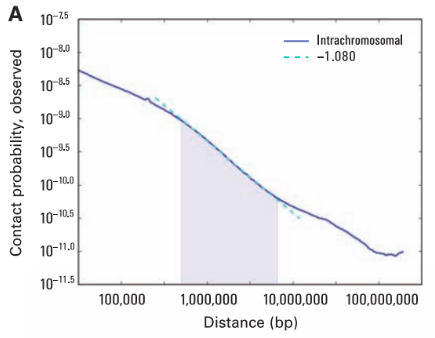
Lieberman-Aiden, 2009
Вероятность контакта хроматина
между двумя участками
зависит от геномного расстояния между ними
P(s)-график
log-log P(s)-график
Когда Hi-C карты не подчиняются закону P(s)?
Oddes 2018
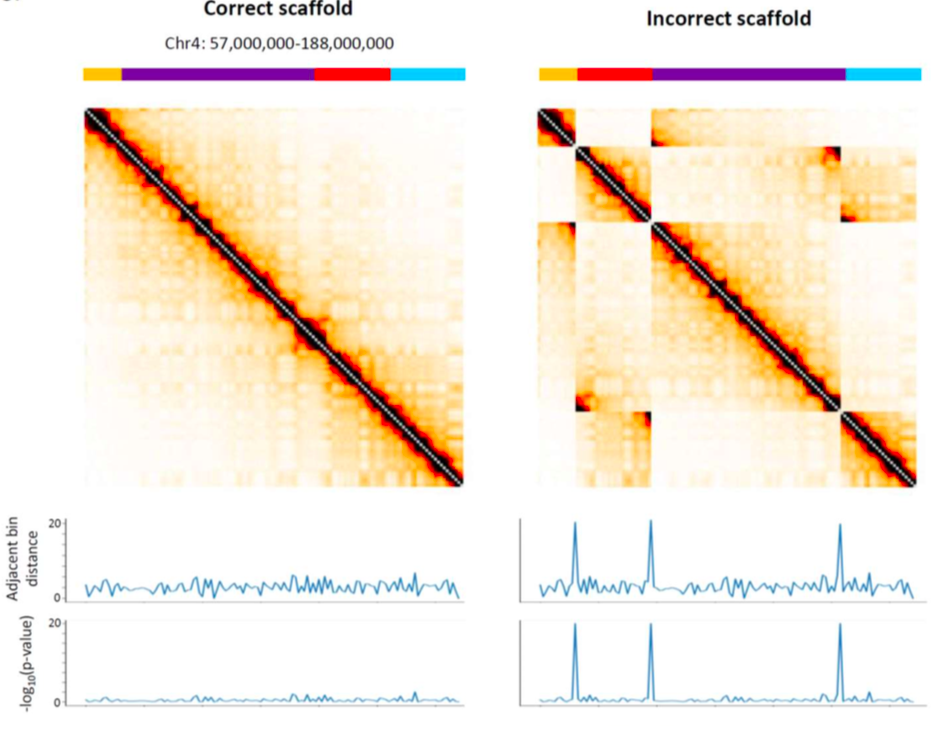
Закон P(s) нарушен?
Или что-то пошло не так с Hi-C?
Правильная последовательность ДНК (сборка)
Ошибка сборки: ДНК устроена не так, как мы думали!
Решение задачи правильной сборки
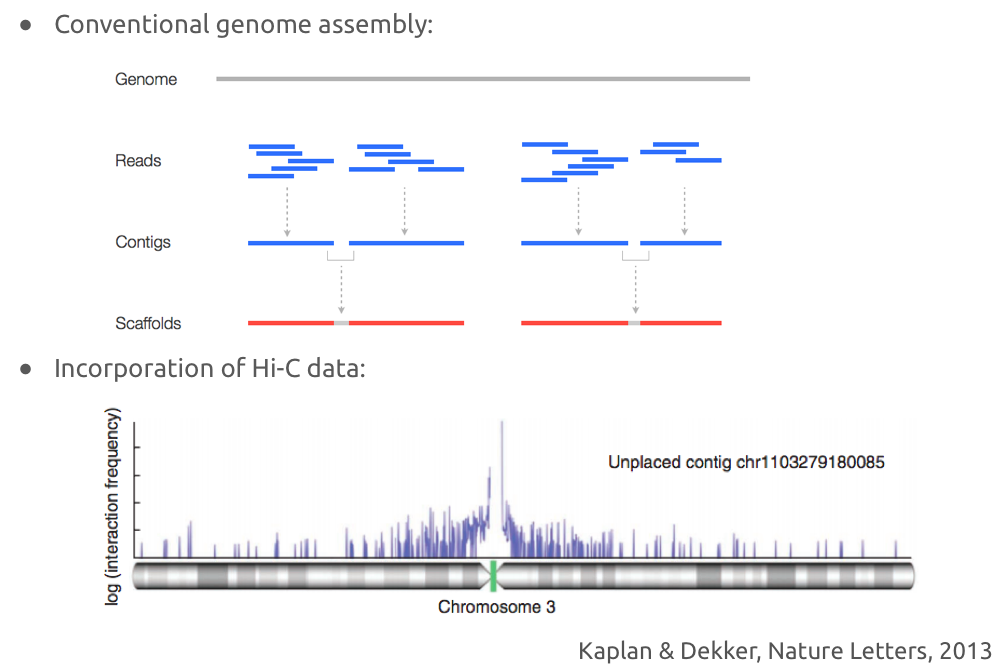
Обычная сборка генома по ридам ДНК:
Улучшение сборки с помощью Hi-C:
Пример успешного решения задачи сборки с помощью Hi-C:
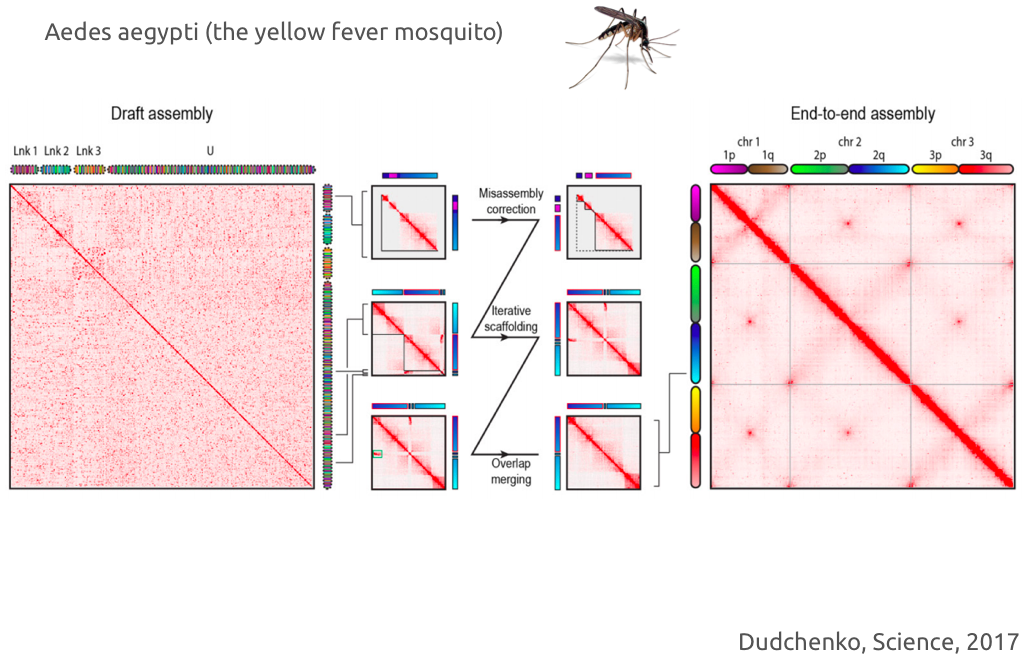
Cборка геномов разнообразных организмов с помощью Hi-C
Обработка данных Hi-C
0. Контроль качества данных NGS
- Программа: fastqc
- Выдача: html-репорт
1. Картирование данных парноконцевого NGS
- Программа: bwa
- Выдача: sam-файл
2. Получение пар контактов Hi-C и их дедупликация
- Программа: pairtools
- Выдача: pairs-файл
3. Бинирование и нормализация
- Программа: cooler
- Выдача: cool-файл
4. Контроль качества
- Программа: multiqc
- Выдача: html-репорт
"Зоопарк"
в хроматиновой лаборатории
Командное задание
Вы работаете в биоинформатической лаборатории по исследованию эволюционных процессов в хроматине. Вам даны:
- данные Hi-C для какого-то организма
(data_1.fastq и data_2.fastq), -
предварительная версия сборки его генома
(genome.fa.gz и chomosomes.sizes.txt).
Ваши задачи:
1) провести процедуру обработки данных;
2) определить организм;
3) вынести вердикт: хорошо ли сработал протокол Hi-C и секвенирование, требуется ли пересборка генома.
Командное задание
Вы работаете в команде, и Вам достаточно выполнить только часть обработки Hi-C, чтобы разобраться.
Вот доступные позиции в лаборатории:
| Роль | |
|---|---|
| Специалист по данным NGS | Контроль качества данных NGS |
| Специалист по картированию | Картирование данных Hi-C |
| Специалист по качеству Hi-C | Контроль качества данных Hi-C |
| Специалист по хроматину | Анализ контактных карт |
| Специалист по биологии | Анализ видовой принадлежности и свойств генома |
Командное задание
Команда состоит из <= 5 участников и получает один набор данных. Один участник может выполнять несколько ролей, но никакие два участника не могут иметь одинаковый набор ролей.
В итоговом отчете Вам нужно:
- указать результаты вашей команды
- подробно ответить на вопросы, находящиеся в сфере компетенции выбранной роли
В суммарный балл входят:
- полнота и научная точность ответа на вопросы для выбранной роли
- командная работа (оценивается по тому, насколько ответы на не-специальные вопросы согласованы с командой)
Руководство к анализу Hi-C
Заходим на кластер
1. Логин через терминал:
или Putty для Windows.
(инструкции: https://www.ssh.com/ssh/putty/windows/#sec-Configuration-options-and-saved-profiles)
2. Активируйте удобный bash-терминал:
3. Создайте директорию проекта:
4. Проверьте, что рабочая среда настроена:
$ ssh -p5222 username@92.242.58.92 или ssh -p32222 username@92.242.58.92$ mkdir practice_chromatin
$ cd practice_chromatin$ bwa -help$ bashСкопируйте данные к себе
- Данные расположены по адресу:
- Выберите номер подпапки по номеру команды:
/usr/share/data-minor-shared/input_data/$ ls /usr/share/data-minor-shared/input_data/team00/В папке содержится информация:
1. Прочтения секвенирования, первые и вторые из пар:
data_1.fastq
data_2.fastq
2. Файл с размерами хромосом данного организма:
chomosomes.sizes.txt
3. Индекированный геном организма:
genome.fa.gz
genome.fa.gz.amb
genome.fa.gz.ann
genome.fa.gz.bwt
genome.fa.gz.pac
genome.fa.gz.sa
Контроль качества данных NGS
$ fastqc ${yourfile.fastq}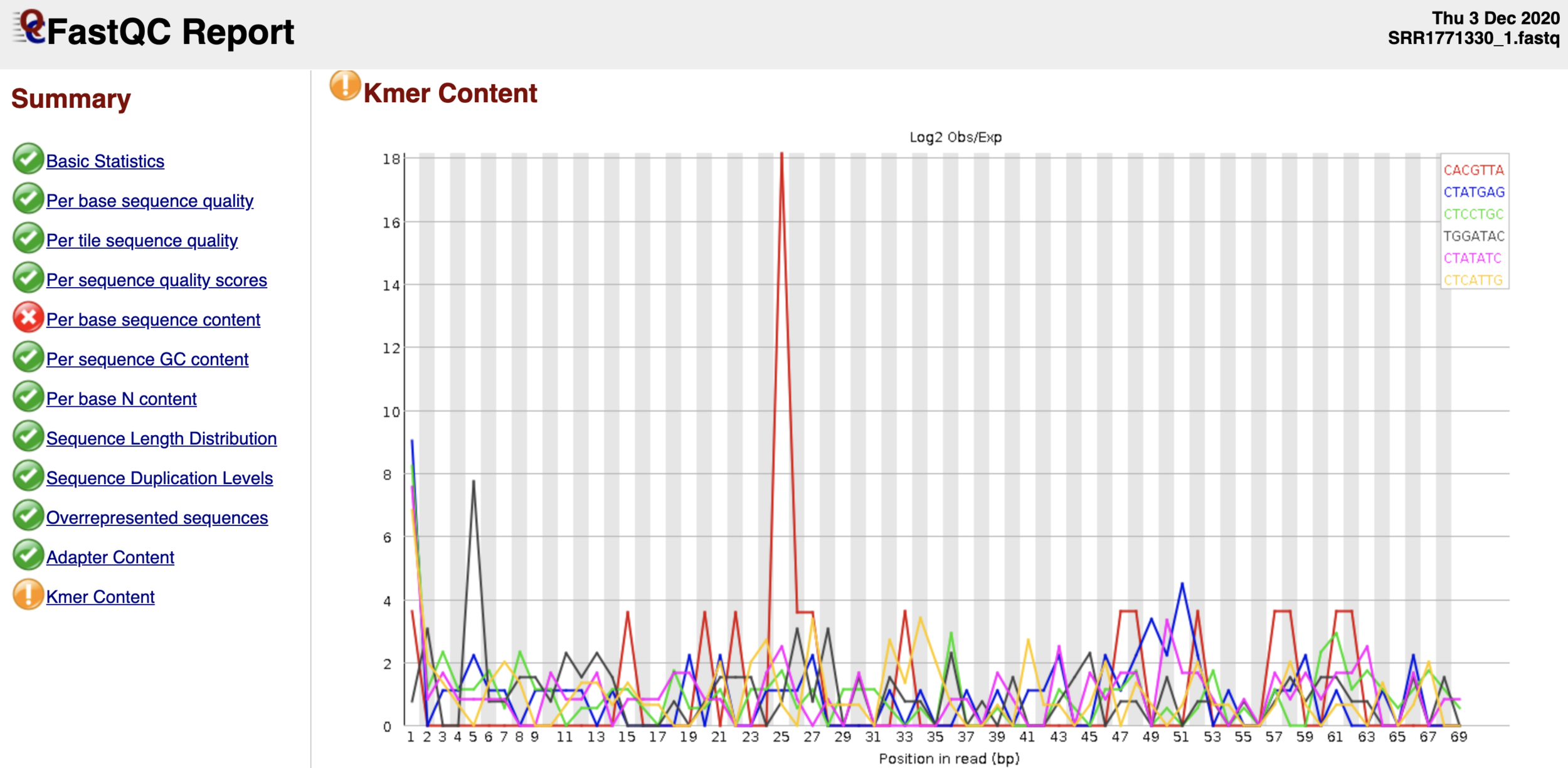
- Запустим fastq и посмотрим на html-репорт (нужно скачать):
(*) в фигурных скобках указано имя файла, которое можно заменить на свое
Картирование данных
- Картирование, переводит
fastq (файл с ридами) в sam (файл с выравниваниями):
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam}$ pairtools parse -c ${chromosome_sizes_file} ${input.sam} -o ${output.pairs} \
--drop-seq --drop-sam --drop-readid --min-mapq 30- Чтение sam в pairs (файл с парами контактов pairs):
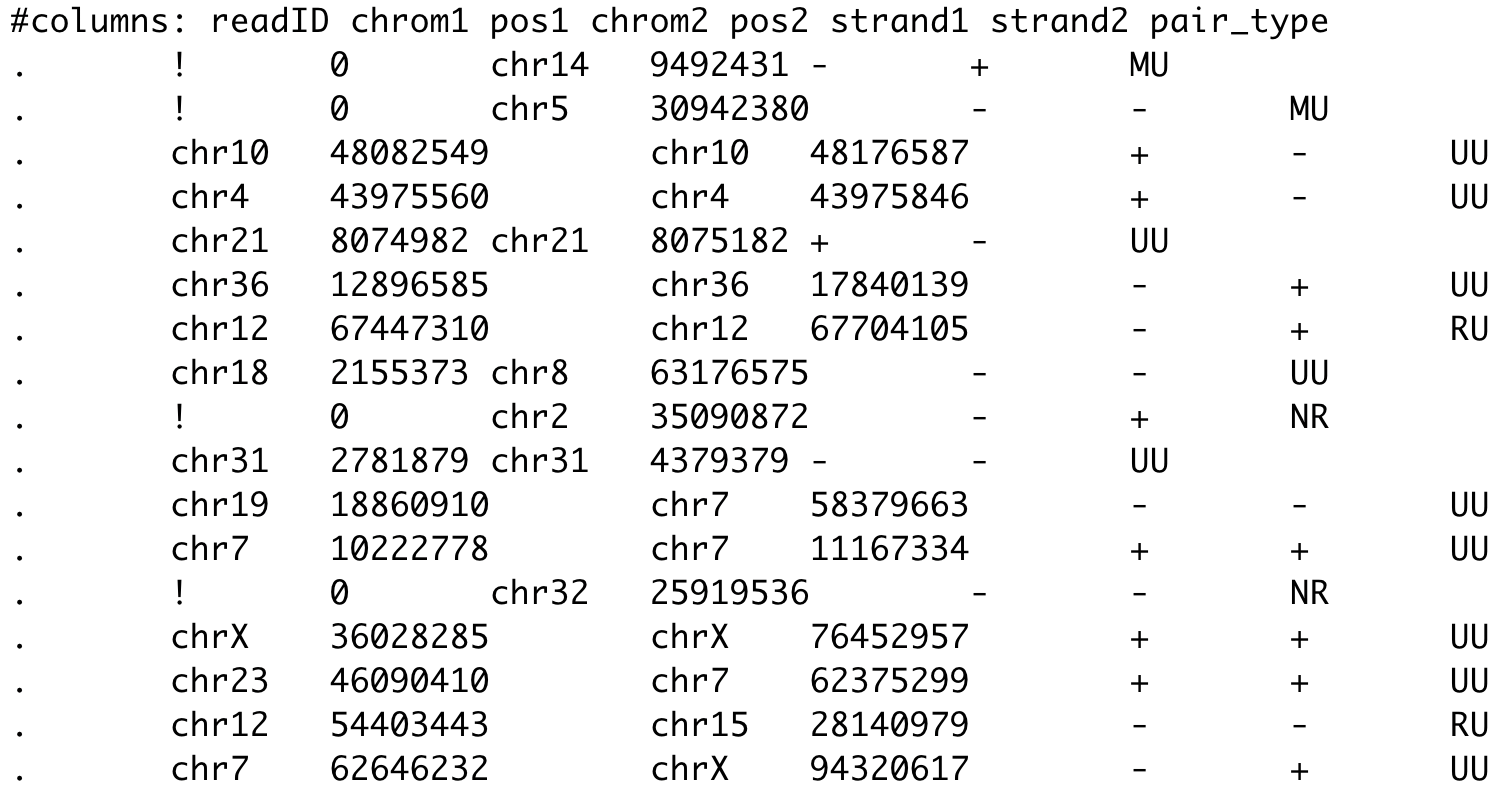
Картирование данных
- Картирование, переводит
fastq (файл с ридами) в sam (файл с выравниваниями):
$ bwa mem -t 1 ${genome_file.fa.gz} ${fastq_file1} ${fastq_file2} > ${output.sam}$ pairtools parse -c ${chromosome_sizes_file} ${input.sam} -o ${output.pairs} \
--drop-seq --drop-sam --drop-readid --min-mapq 30- Чтение sam в pairs (файл с парами контактов pairs):
$ pairtools dedup --output-stats ${output.dedup.stats} ${output.pairs} \
-o ${output.nodups.pairs}- Дедупликация pairs-файла:
Контроль качества пар
- Запуск multiqc на выдаче описательной статистики:
multiqc --module pairtools ./- Файл с описательной статистикой был на предыдущем шаге:
$ pairtools stats -o ${output.stats} ${output.nodups.pairs}Контроль качества Hi-C

Типы картирований:
Контроль качества пар
Разные типы пар контактов:
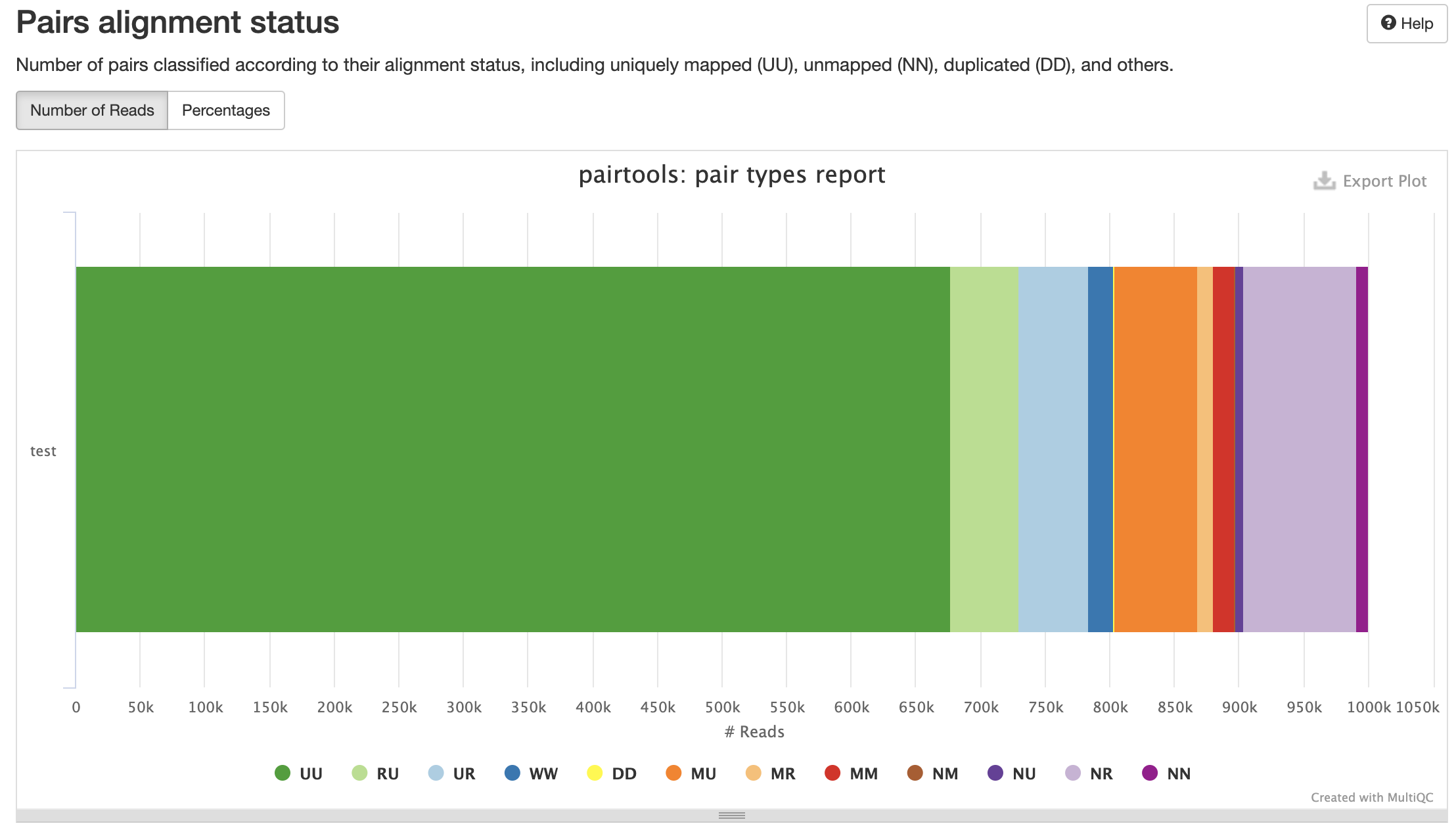
Контроль качества пар
Количество контактов для разного геномного расстояния:
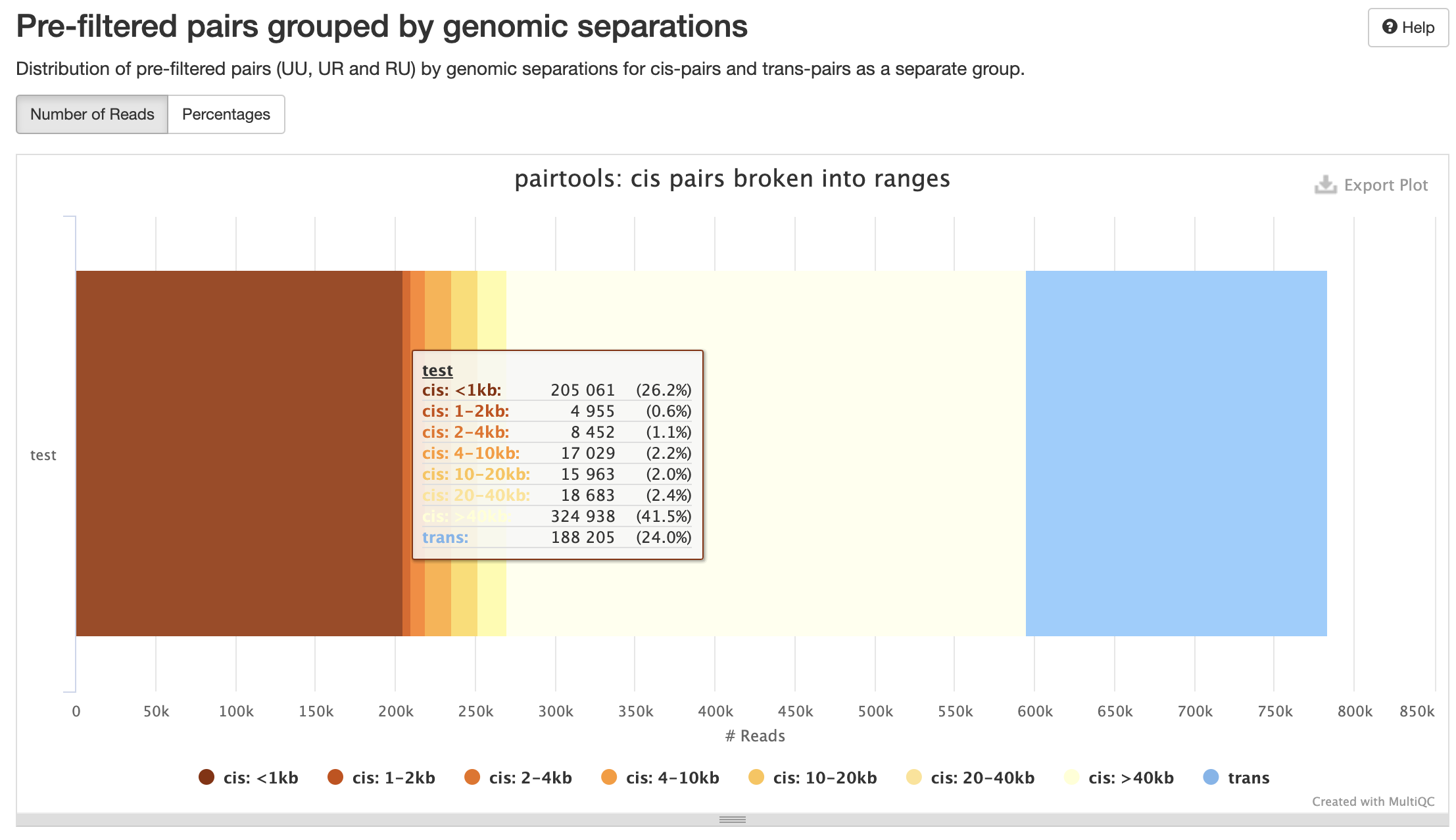
Контроль качества пар
P(s):
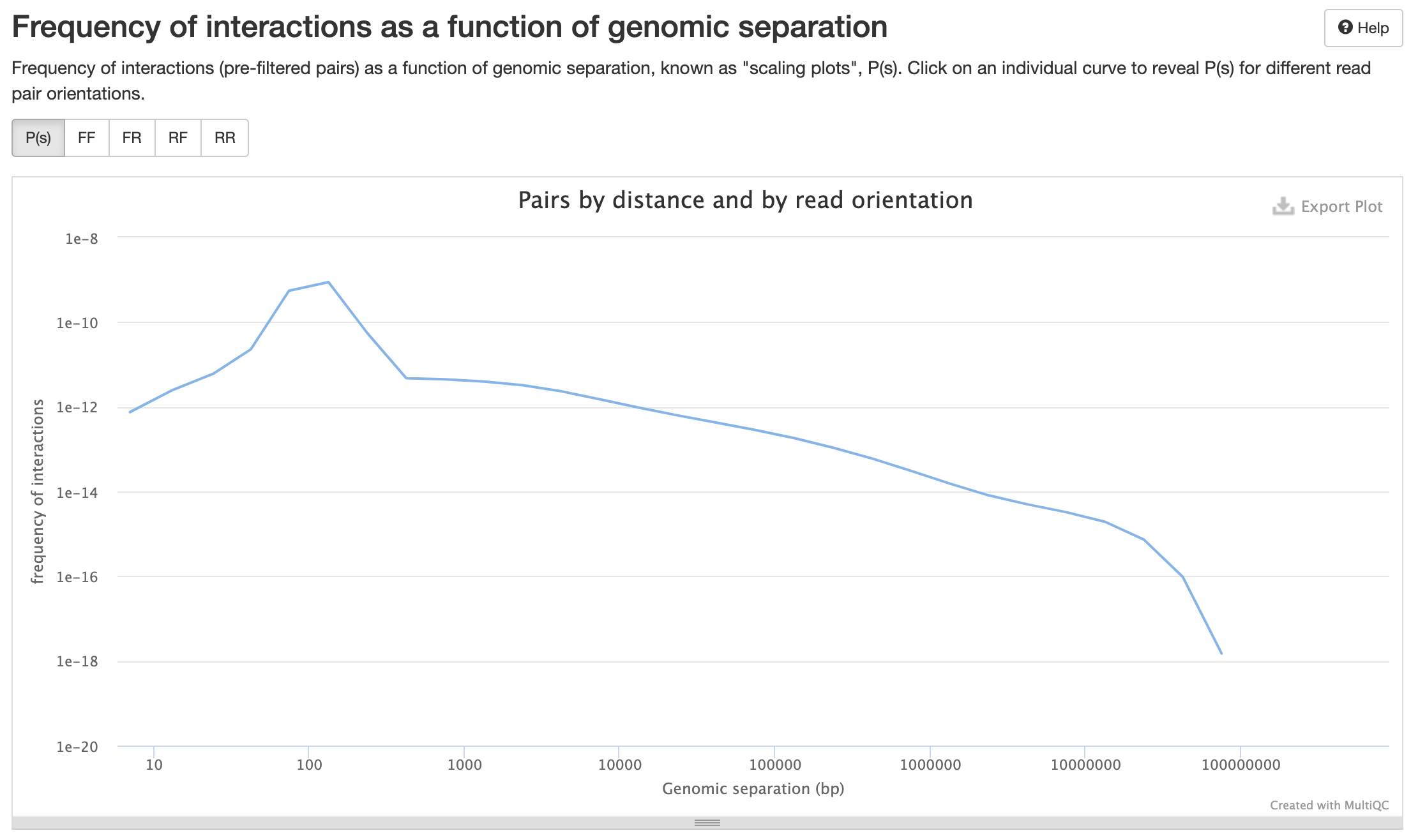
Получение контактных карт
$ cooler cload pairs -c1 2 -c2 4 -p1 3 -p2 5 ${chromsizes}:1000000 ${input.nodup.pairs} ${out.cool}- Загрузка контактных пар в cool-файл:
$ cooler balance ${out.cool}- Проводим корректирование Hi-C данных:
$ cooler info ${out.cool}- Проверяем запись и информацию:
(*) 100000 - это размер одного бина Hi-C карты, иначе говоря, 100 Kb
Визуализация карт
$ cooler show ${in.cool} chr1 -o ${image.png}- Просмотр одной хромосомы:
- Для просмотра полного генома воспользуйтесь HiCExplorer:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicPlotMatrix.html#hicplotmatrix
Подсказки:
- Для определения вида организма, воспользуйтесь тем, что вам известно, какие риды закартировались на его геном. Откройте sam-файл для просмотра и выберите последовательности закартировавшихся регионов. Запустите на них BLAST.
- Для получения информации о хромосомах откройте файл с размерами хромосом для просмотра. Для подсчета количества хромосом/контигов можно воспользоваться командой wc -l.
- Информацию о сборке генома исследуемого организма (после его определения) можно найти на сайте NCBI.
Цикл лекций о структуре хроматина: Практическая работа с Hi-C
By agalicina




