Reproducible data analysis with Nextflow & NF-core
Alexander Peltzer
Quantitative Biology Center (QBiC) Tübingen

Outlook
- Challenges in computational biology
- Basic principles of Nextflow
- Introduction to NF-core project
Challenges: Big Data
- Data in computational biology is
- big (PB scale)
- diverse (sequencing, proteomics, metabolomics ...)
- erroneous (e.g. contains sequencing errors)
We need methods and tools to analyze such data!
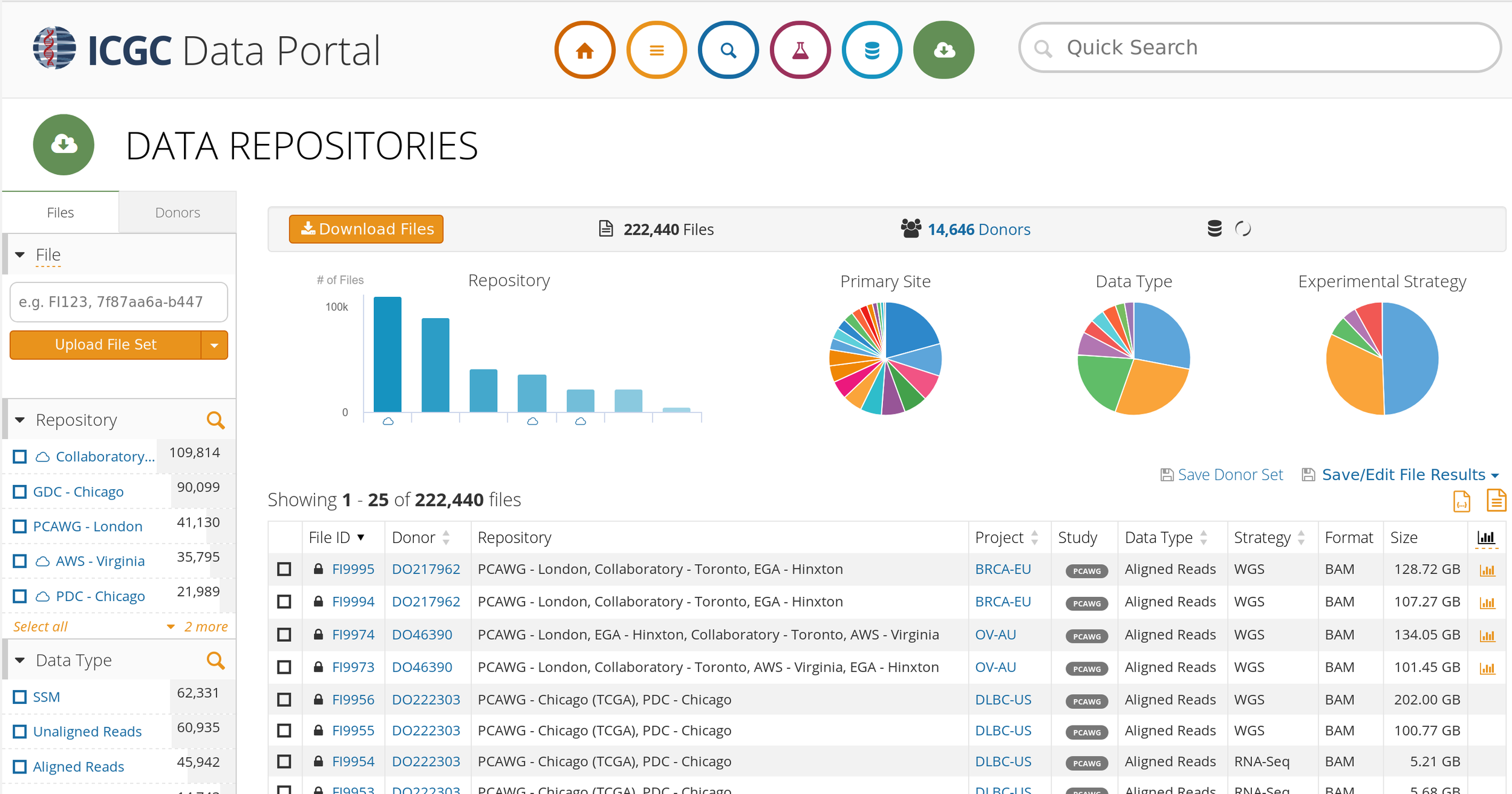
Challenges: Big Data - ICGC
Text
"Hyper-Moore gap"
Text

Credit to Swaine Chen, Genome Institute of Singapore, AWS Summit 2018
The FAIR* principle
Findable
Accessible
Interoperable
Reproducible
The FAIR Guiding Principles for scientific data management and stewardship, Wilkinson et al. 2016 qPortal: A platform for data-driven biomedical research, Mohr et al. 2018
DOI
qPortal
?
?
Challenges: Software dependencies
Workflows / Pipelines consist of
- different tools
- dozens of individual methods
Complex dependency trees and configuration requirements!
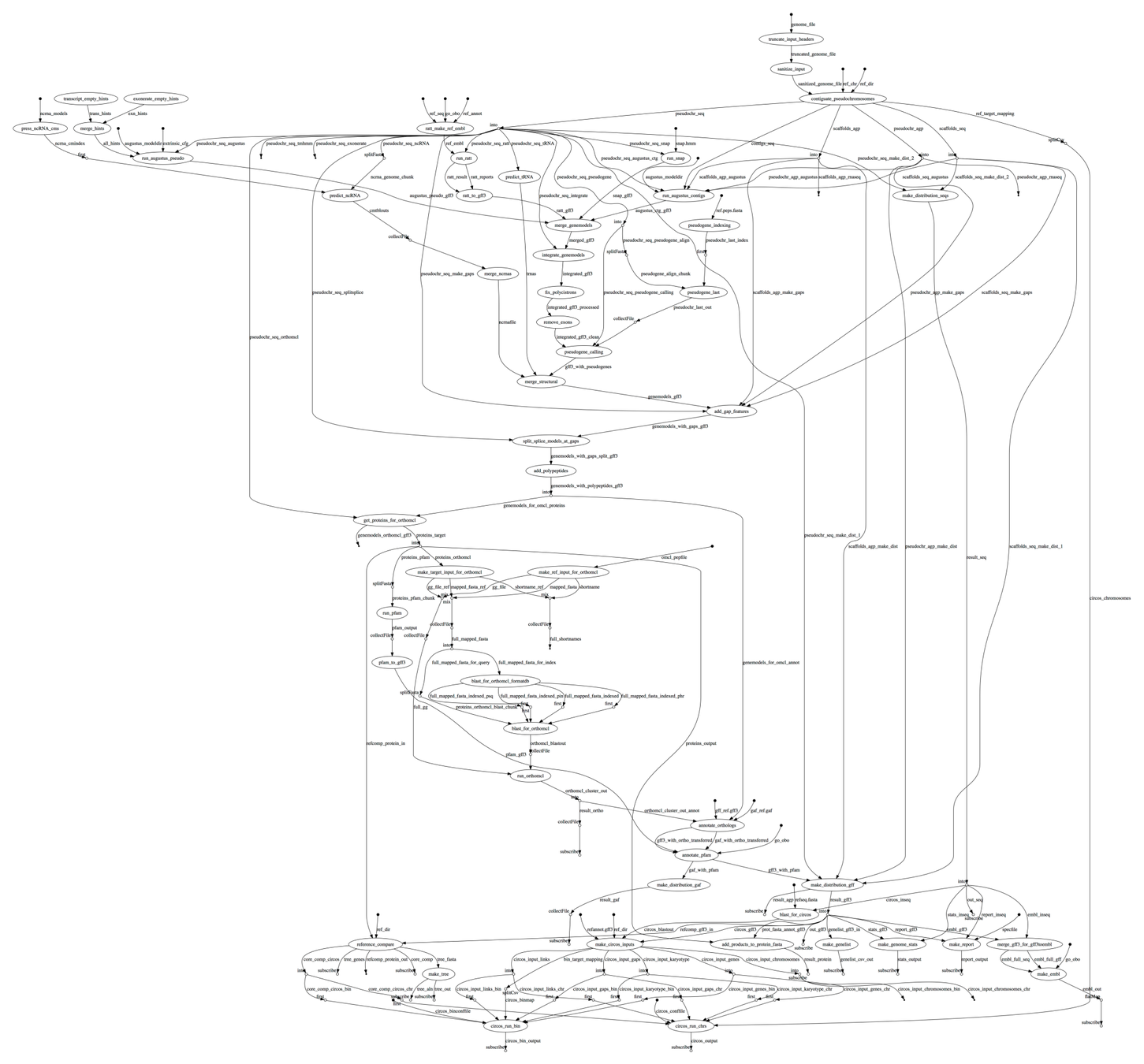
Steinbiss et al., "Companion: a web server for annotation and analysis of parasite genomes", NAR 2016
Challenges: Reproducibility
- Large-scale projects more common today
- 1,000 Genomes Project
- 100,000 Genomes Project UK
- Reproduce results with older data / integrate with newer data
- Challenging: Many paper results are not reproducible at all or require a lot of effort
Challenges: Reproducibility

"We estimated the overall time to reproduce the method as 280 hours for a novice with minimal expertise in bioinformatics."
Challenges: Environmental stability
- Portability and stability of code between different OS should be ensured
- Are results different? Yes, they are ...
Challenges: Software dependencies
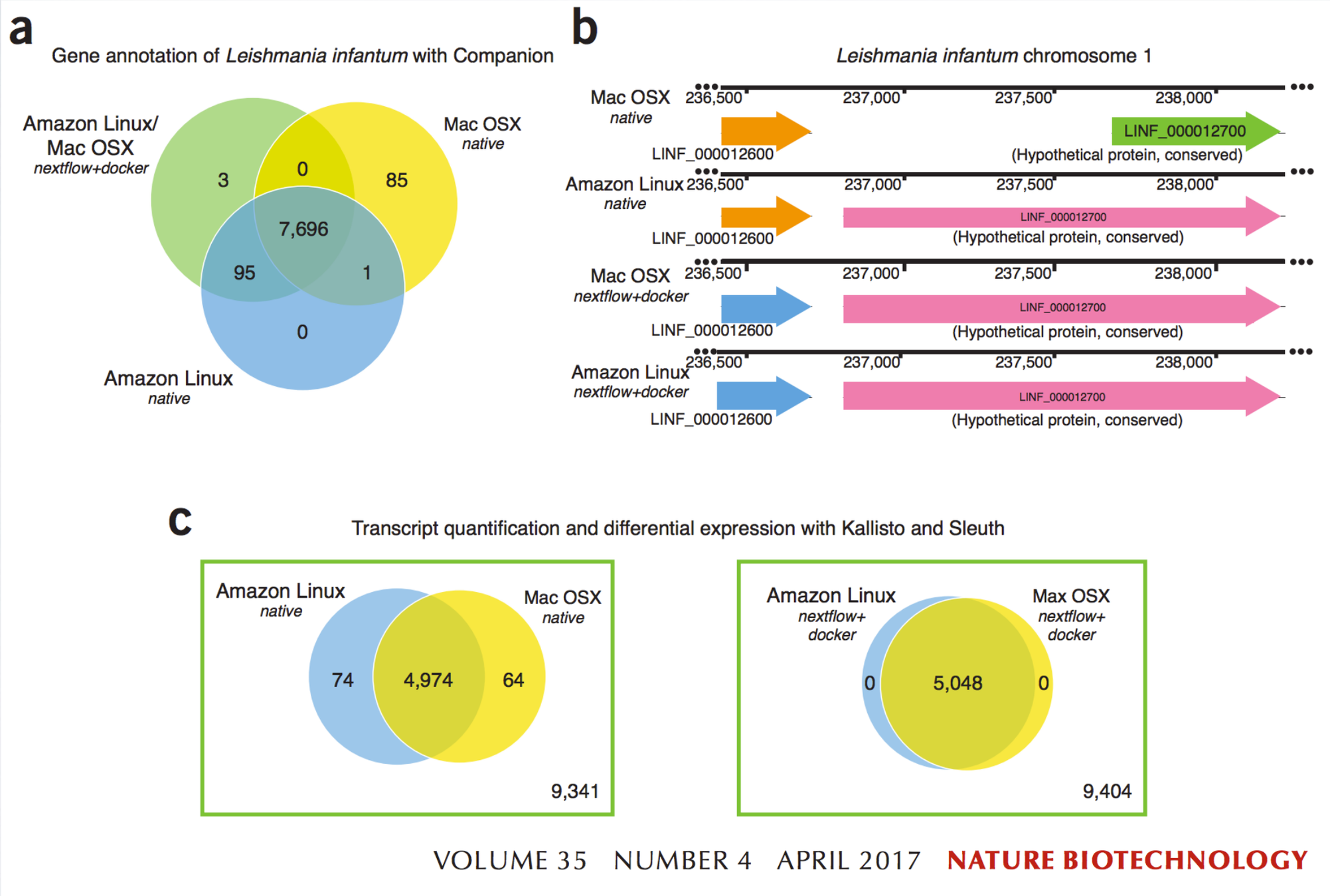
diTommaso et al., 2017, Nature Biotechnology
Nextflow
- Custom DSL (domain-specific language) for
- fast prototyping
- enabling task composition
- easy parallelization
- Self-contained: Containerize tasks (e.g. with Docker)
- Isolation of dependencies: Keep container - rerun analysis at any point!
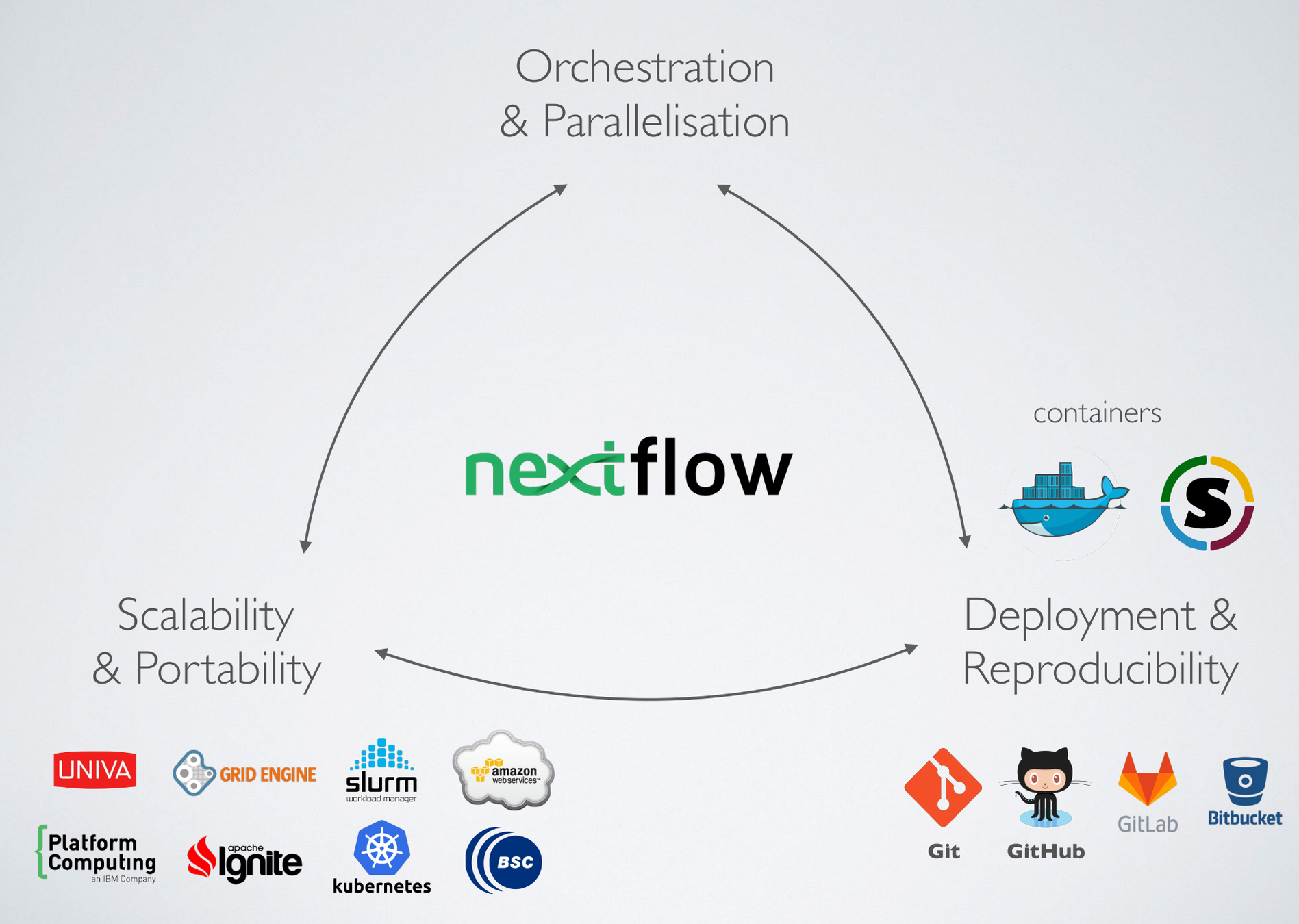
(credit to E Floden, CRG Barcelona)
Nextflow: Centralised Orchestration
Nextflow
Cluster
- Submit jobs to cluster nodes
- Store data on shared storage
Storage
Nextflow: Cloud deployment (AWS)
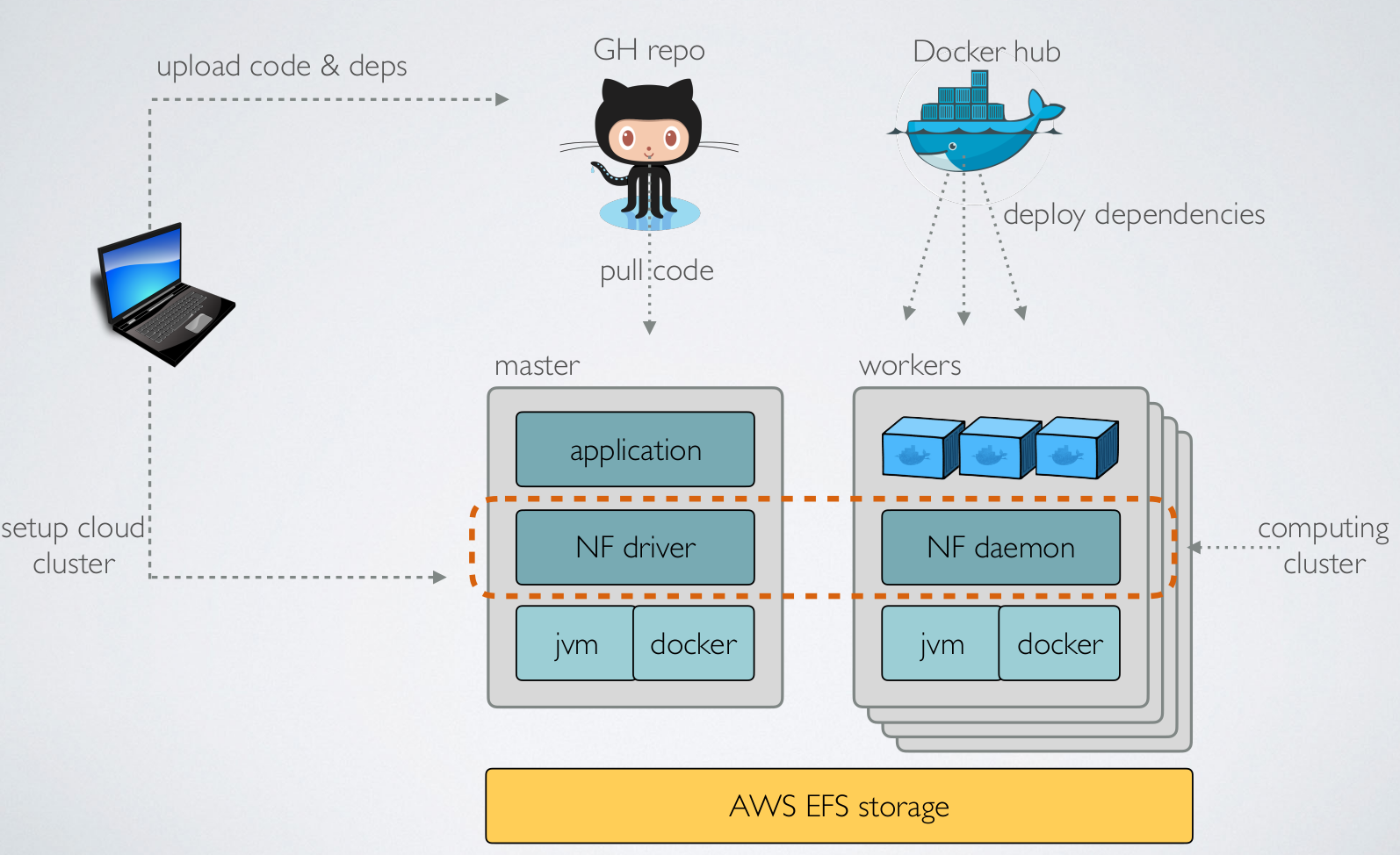
(credit to E Floden, CRG Barcelona)
Platform support

(credit to E Floden, CRG Barcelona)
Nextflow: Executor abstraction
Improves code portability
#Run me locally
process.executor = 'local'
#Run on AWS Batch
process.executor = 'awsbatch'
#Run on Kubernetes cluster
process.executor = 'k8s'
- Community effort to collect production ready analysis pipelines
- Save time in development, more testing, more updates
- https://nf-co.re





Phil Ewels
Alex Peltzer
Sven Fillinger
Andreas Wilm
Maxime Garcia
+ many others!

Tiffany Delhomme
- Community effort to collect production ready analysis pipelines
- Save time in development, more testing, more updates
- Initially supported by SciLifeLab, QBiC and A*Star Genome Institute Singapore


All pipelines adhere to requirements
- Nextflow based
- MIT license
- Software bundled in Docker
- Continuous integration testing (e.g. Travis CI)
- Stable release tags
- Common pipeline usage and structure
Dockerfiles
FROM nfcore/base
MAINTAINER Phil Ewels <phil.ewels@scilifelab.se>
LABEL authors="phil.ewels@scilifelab.se" \
description="Docker image containing all requirements for the nfcore/rnaseq pipeline"
COPY environment.yml /
RUN conda env create -f /environment.yml && conda clean -a
ENV PATH /opt/conda/envs/nfcore-rnaseq-1.5dev/bin:$PATHDockerfiles
- Bioconda package based, clean-style
name: nfcore-rnaseq-1.5dev
channels:
- bioconda
- conda-forge
- defaults
dependencies:
- conda-forge::openjdk=8.0.144
- fastqc=0.11.7
- trim-galore=0.4.5
- star=2.6.0c
- hisat2=2.1.0
- picard=2.18.7
- bioconductor-dupradar=1.8.0
- conda-forge::r-data.table=1.11.4
- conda-forge::r-gplots=3.0.1
- bioconductor-edger=3.20.7
- conda-forge::r-markdown=0.8
- preseq=2.0.3
- rseqc=2.6.4
- samtools=1.8
- stringtie=1.3.4
- subread=1.6.1
- multiqc=1.5Docker/Singularity processes
- Mounting paths is done via Nextflow
- No tuning for LustreFS/GPFS necessary
In general: Seamless integration of resources outside of containers - without further required changes!
Docker/Singularity processes
- Mounting paths is done via Nextflow
- No tuning for LustreFS/GPFS necessary
In general: Seamless integration of resources outside of containers - without further required changes!
Docker/Singularity processes
- Mounting paths is done via Nextflow
- No tuning for LustreFS/GPFS necessary
In general: Seamless integration of resources outside of containers - without further required changes!
Docker/Singularity build processes
- Automated via TravisCI
- Release TAG on GitHub => Release on Travis CI => Push to DockerHub/SingularityHub
Optional requirements
- Software bundled in bioconda
- Optimised output formats (e.g. CRAM)
- Explicit support for cloud environments (AWS)
- Benchmarks for running on such environments
Need help?
- Cookiecutter: To get a skeleton for new pipelines
- Linting app: To check what conforms with nf-co.re
- Gitter: To communicate with the community!
It's demo time!
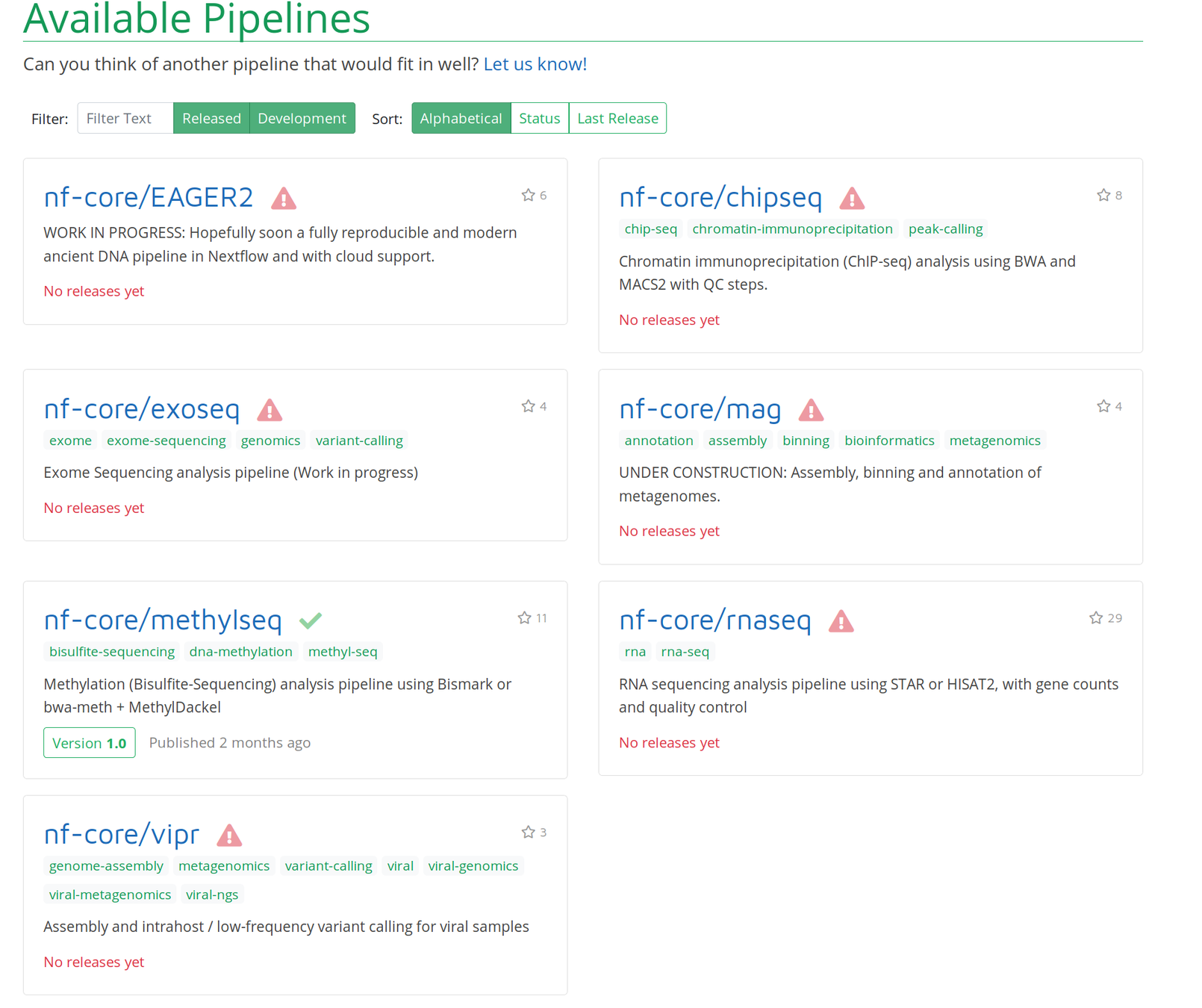
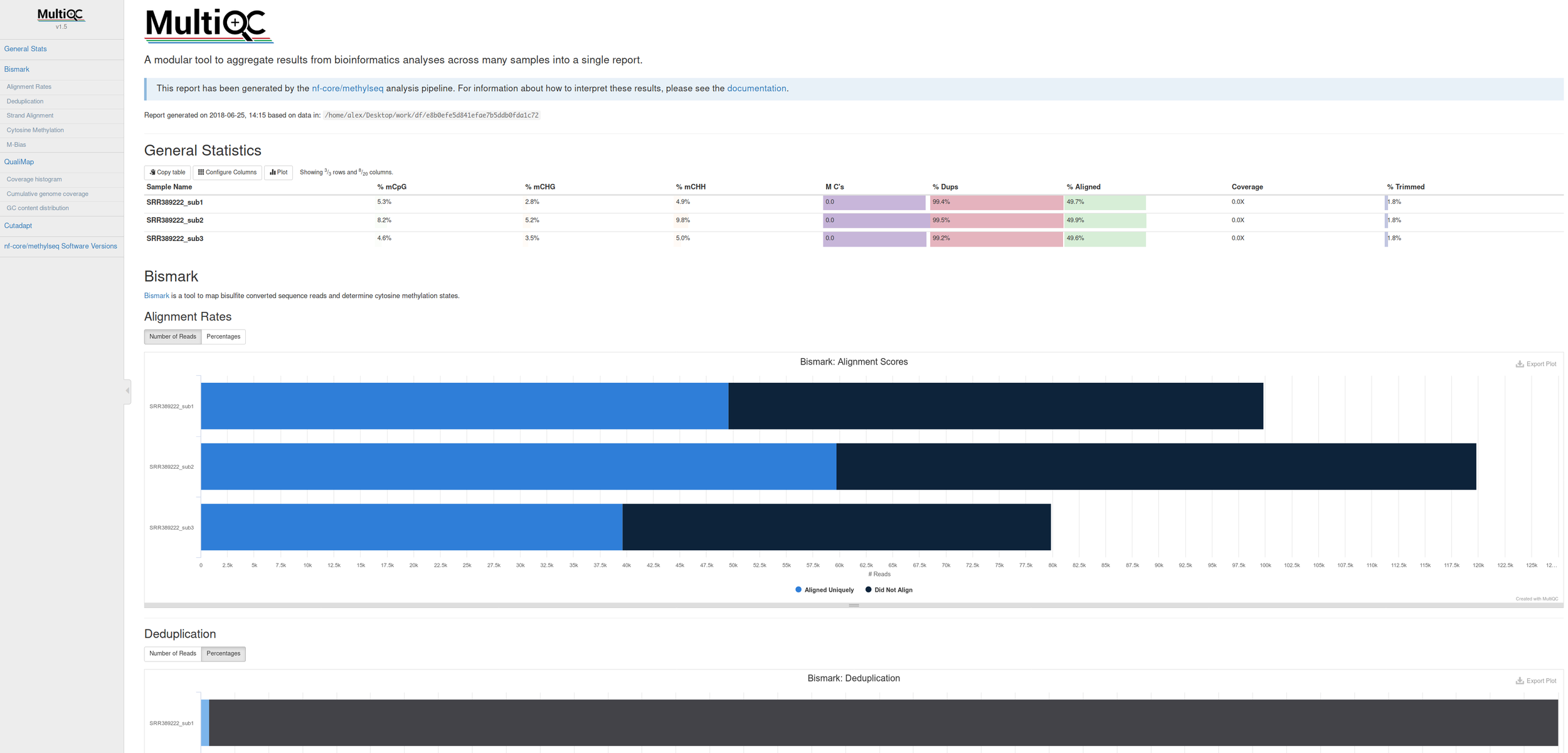
Comes with interactive reports!
Comes with proper documentation!
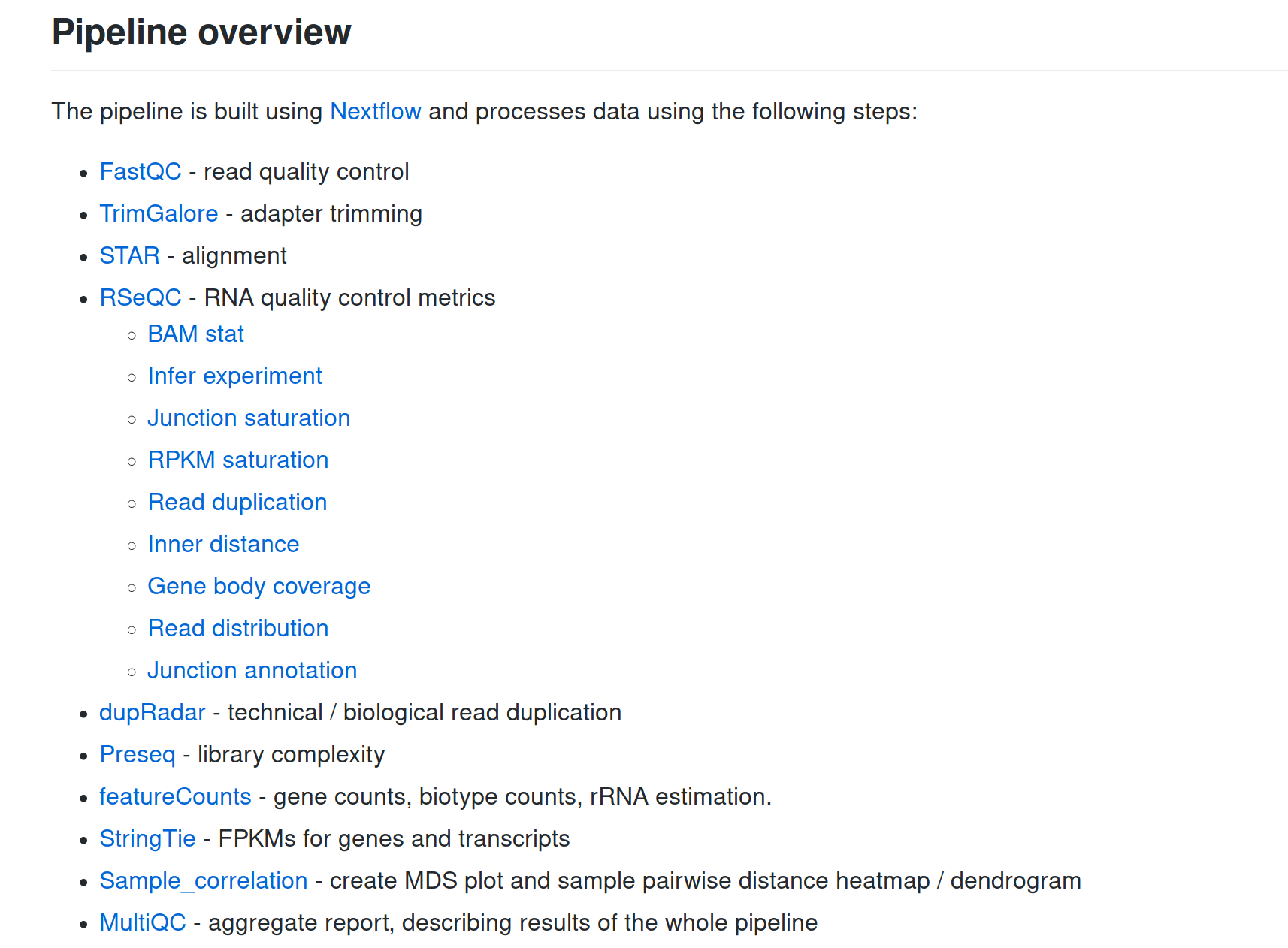
... and a lot more!

Acknowledgements
Phil Ewels (SciLifeLab)
Maxime Garcia (SciLifeLab)
Sven Fillinger (QBiC)
Paolo di Tommaso (CRG)
Evan Floden (CRG)
Andreas Wilm (A* Singapore)
2018-07-17_S+C_NF-Core
By Alexander Peltzer
2018-07-17_S+C_NF-Core
NF Core mini talk for presentation at AtoS / S+C in Tübingen on July 17, 2018.



