Calibration of amino acids for constant-pH simulation
Bas Rustenburg // Chodera lab
#pHallthetime
4/20/2016
Disclaimer
For the purpose of this presentation, I've experimented with multiple ways to provide a more interactive experience. Feedback is welcome.
Follow along online!
Follow the slides live on your own computer using this url:
#pHallthetime
http://slides.com/
basrustenburg/aminoallthetime/live
Why should I care about constant-pH simulations?
Protonation state effects
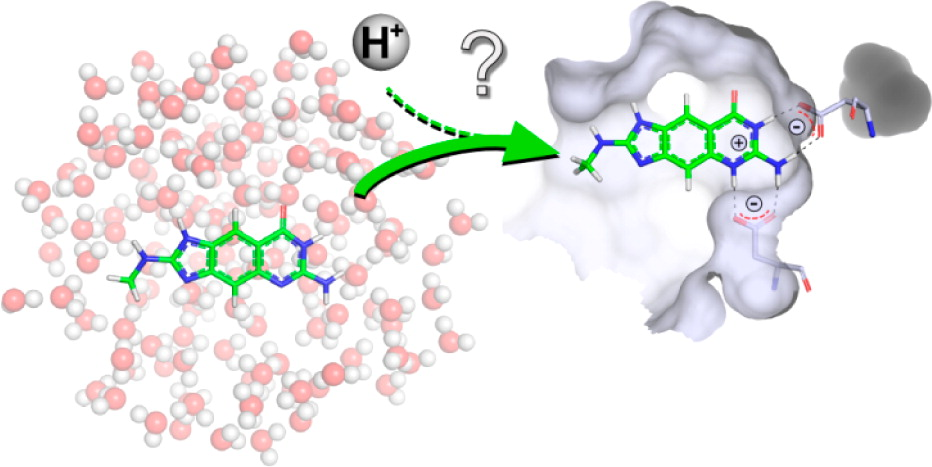
Neeb et al. J. Med. Chem., 2014, 57 (13), pp 5554–5565
There is no a priori way to know every relevant protonation state of a ligand or protein, and how relevant they are to the binding affinity.
The standard state approach
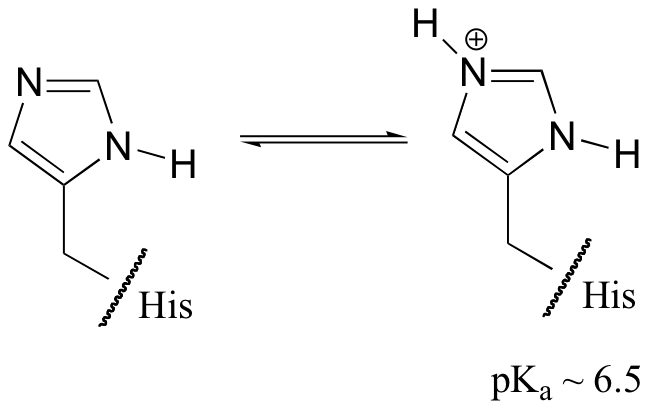
- Typically, one only simulates a single protonation state.
- For proteins at neutral pH, only histidine is closely looked at.
Problems
- If reality contains a mixture of protonation states, we're ignoring their contributions to the free energy of the system
- If the most relevant protonation state changes upon binding, we simulate the wrong state
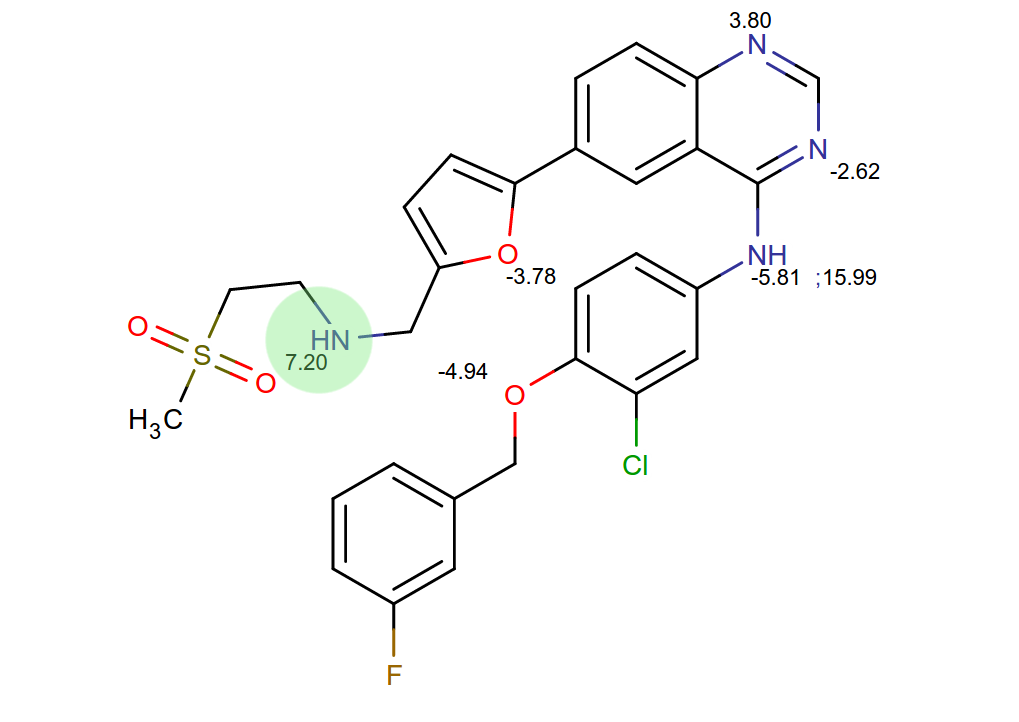
The standard approach is ignoring contributions of unknown magnitude, for imatinib and Abl there is clear indication of the relevance
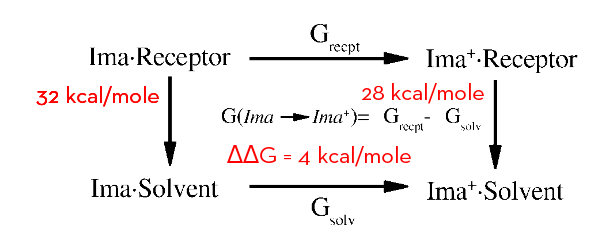
Aleksandrov, A and Simonson, T J Comput Chem 31,7, pp. 1550–1560 (2010)
More problems with the Standard aproach
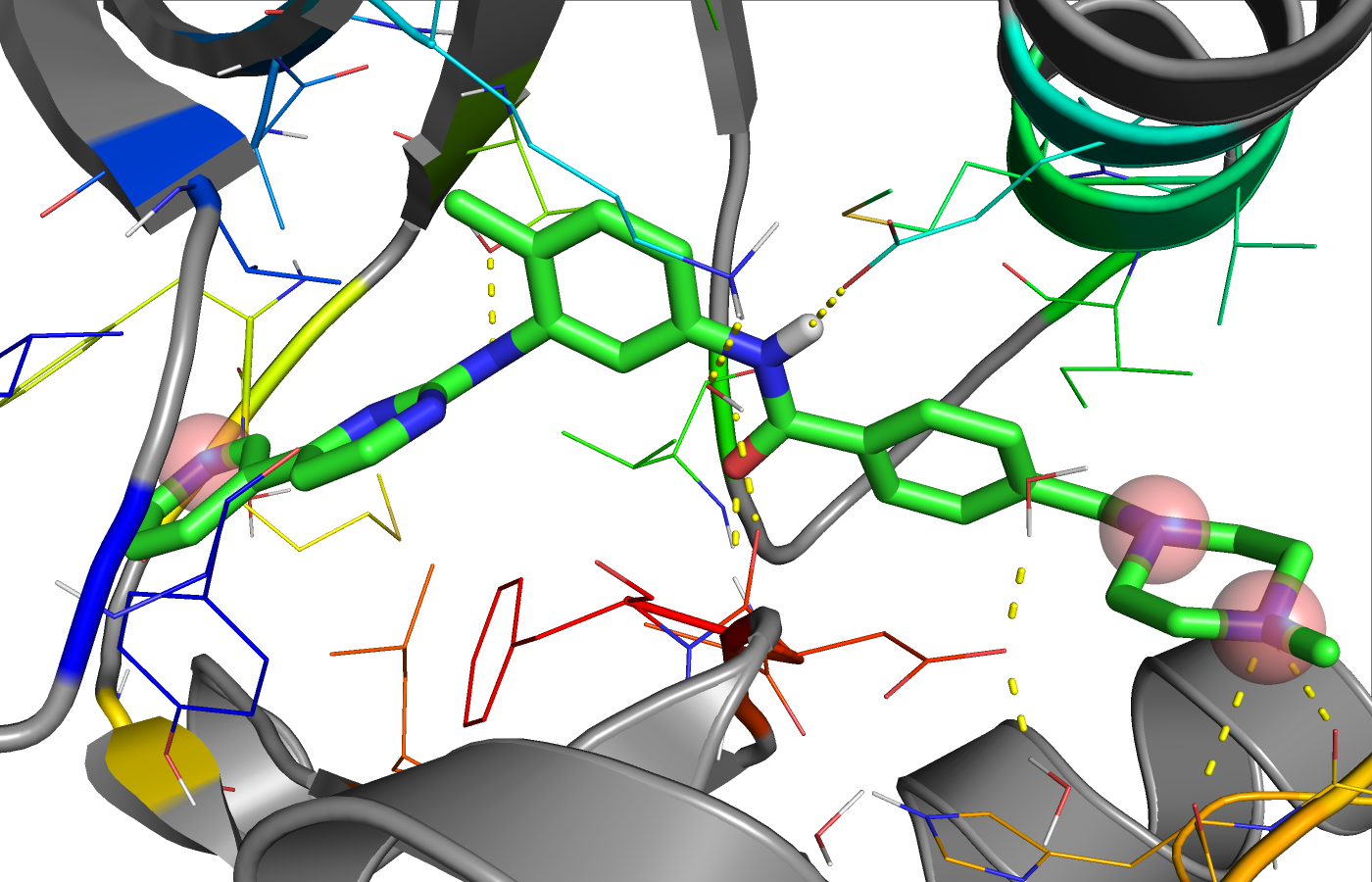
Imatinib bound to Abl kinase
Kinase inhibitors
pKa predictions using EPIK suggest that many kinase inhibitors have more than one accessible protonation state at pH 7.4

Figure by: John & Julie
How does constant-pH
solve our problems?
#pHallthetime
Constant PH
Allow for the exchange of protons with an external proton "bath", which maintains a constant pH.
a_{H^+} = \exp \left( \frac{\mu_{H^+} - \mu^\ominus_{H^+} } {RT} \right)
pH = - \log_{10} (a_{H^+}) \approx - \log_{10} [H^+]
#pHallthetime
Mongan et al. Constant pH Method
In implicit solvent
- Take a reference state with a known pKa, such as a free dipeptide or capped peptide in solution
- ΔGelec difference between the electrostatic potential for the current protonation state and proposed state
- ΔGelec,ref the free energy difference in a reference state (solvent)
\Delta G = k_{\mathrm{B}}T \color{cyan}{\left(\mathrm{pH - pK_{a,ref}} \right) \ln 10 } + \color{red}{\Delta G_{\mathrm{elec}}} - \color{orange}{\Delta G_{\mathrm{elec,ref}}}
Mongan, Case, and McCammon • Journal of Computational Chemistry
Volume 25, Issue 16, pages 2038–2048, December 2004
Constant PH
In implicit solvent
- (ΔGelec) take difference between the electrostatic potential for the current protonation state and proposed state
- Accept using
\Delta G = k_{\mathrm{B}T} \left(\mathrm{pH - pK_{a,ref}} \right) \ln 10 + \Delta G_{\mathrm{elec}} - \Delta G_{\mathrm{elec,ref}}
Mongan, Case, and McCammon • Journal of Computational Chemistry
Volume 25, Issue 16, pages 2038–2048, December 2004
P =
\begin{cases}
1 & \quad \text{if } \Delta G \leq 0 \\
e^{-\Delta G / k_{BT}} & \quad \text{if } \Delta G > 0
\end{cases}
Issues with the Mongan method
\Delta G = k_{\mathrm{B}}T \color{cyan}{\left(\mathrm{pH - pK_{a,ref}} \right) \ln 10 } + \Delta G_{\mathrm{elec}} - \color{orange}{\Delta G_{\mathrm{elec,ref}}}
Mongan, Case, and McCammon • Journal of Computational Chemistry
Volume 25, Issue 16, pages 2038–2048, December 2004
-
Inefficient/unsuited for explicit solvent
- instantaneous MC
- no counterions
-
Ref. free energies are inaccurate
- Not transferable
- pKa references most likely not appropriate for proteins
How do we get accurate reference energies?
How do we get accurate reference energies?
Or rather, how do we reproduce the experimental histogram of states?
#pHallthetime
Updated scheme
In whatever solvent
- Calculate free energy of according to:
, where
- is calibrated using SAMS
\Delta G_{ij, \text{protonation}} = \Delta G_\text{elec} + \Delta g_\text{ref}
\Delta g_{ref,k}
j \rightarrow i
#pHallthetime
\Delta g_\text{ref} = g_{\mathrm{ref,j}} - g_{\mathrm{ref,i}}
Probability of a protonation state
\log P(x;L\!=\!j) = - \beta \left( \mathcal{H} (x;L) + p V N_A + \Delta G _{\text{ref}j} \right)
- \text{proton count} \cdot (pH - pKref) \cdot \log(10)
Calibration of ref. energy
\log P(x;L\!=\!j) = - \beta \left( \mathcal{H} (x;L) + p V N_A + \zeta_j \right)
- \text{proton count} \cdot (pH - pKref) \cdot \log(10)
We configure the reference state with SAMS
Short recap of SAMS
- Assign target weights to every state/label
- Perform labeled mixture sampling of
- Compute weights using update scheme
k, \pi_k
L_t, X_t
\zeta^{(t)} = \zeta^{(t - \frac{1}{2})} - \zeta_{1}{\!}^{(t-\frac{1}{2})}
\zeta_j = \ln{ \frac{Z_j}{Z_1}}
Calibration of ref. energy
\log P(x;L\!=\!j) = - \beta \left( \mathcal{H} (x;L) + p V N_A + \zeta_j \right)
- \text{proton count} \cdot (pH - pKref) \cdot \log(10)
We want to replace this:
Definition of gk
\log P(x;L\!=\!j) = - \beta \left( \mathcal{H}(x;L) + p V N_A + g_k \right)
With this:
\log P(x;L\!=\!j) = - \beta \left( \mathcal{H}(x;L) + p V N_A + \zeta_j \right)
- \text{proton count} \cdot (pH - pKref) \cdot \log(10)
Using SAMS to calibrate constant-pH simulations
-
Obtain target weights from pH curve given pKa
- For ligands: Epik proto/tautomer populations
- Regular constant-pH code already samples states and configurations
- Compute weights using update scheme
k, \pi_k
L_t, X_t
g^{(t)} = g^{(t - \frac{1}{2})} - g_{1}{\!}^{(t-\frac{1}{2})}
Sams run for histidine
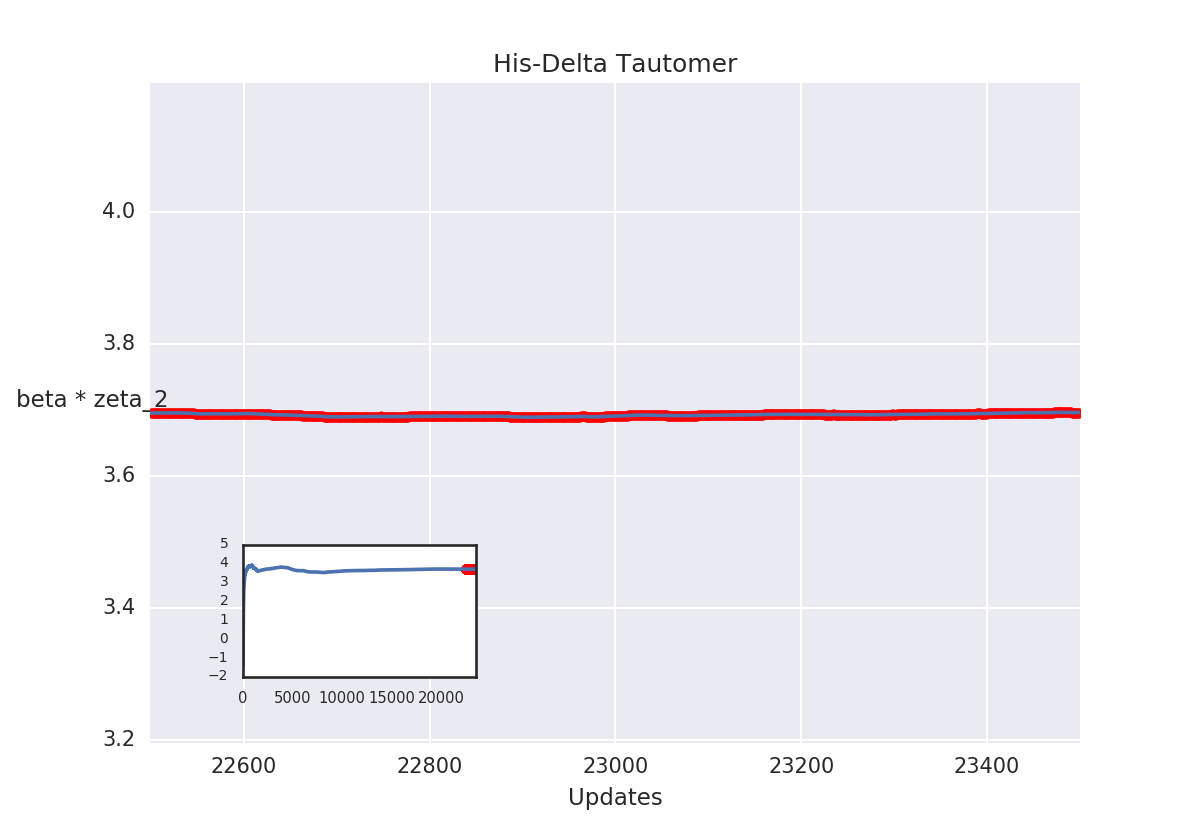
We're not there yet...
- Instead of uniform sampling, we see populations:
- 0: 0.5535535535535535
- 1: 0.27127127127127126
- 2: 0.17517517517517517
How can we increase state overlap?
- We need good overlap between states
- When some states have low it's hard to get overlap
- Can we reweight starting from uniform weights?
\pi_k
How do estimate convergence?
Convergence criterion
- Ideally, we'd have an estimator of the variance
- We'd then run SAMS until our variance is below a certain threshold
- Instead, I decided to use a gradient
- For instance, stop iteration when gradient below
- Using a simple np.gradient code
- Alternate idea:
- Uses the already calculated sams updates
\zeta^{t - \frac{1}{2}}
1 \cdot 10^{-6}
Convergence criterion
Or, perhaps quit when the observed histogram of labels matches the target weights within a given percentage:
p(L = j | x; \zeta) \approx \pi_j
Ways to improve calibration scheme
- Start runs at multiple initial guess values
- How to cleverly decide the closest guess.
- Initially sample uniformly from states
- Converges faster, and gives better initial guess
- Then, change the target weights and optimize.
Explicit solvent considerations
- Using NCMC with SAMS global scheme
- Better overlap between states
- The effect of ion concentrations
- On the free energy
- On convergence
- Expand to incorporate LJ parameters
- Currently, just charges are supported
- Support for ligands
- Coordinated changes (pairs of residues, etc.) to increase acceptance probability
- Pick pairs at random
- Find a way to pick pairs close together that satisfies detailed balance
- Bias picks to low-probability/high free energy cost states
Future plans: ConstpH
Thanks for your attention
Confucius says:
"Transfer some protons today!"
Update Constph
By Bas Rustenburg
Update Constph
8/10/2016 // Gunner lab



