











Chengcheng Xiao
PhD student @ Imperial College London
Basis (PW, local set)
DFT calculator
Bloch WFs
Bloch WFs
???
Basis (PW, local set)
Wannier transformation
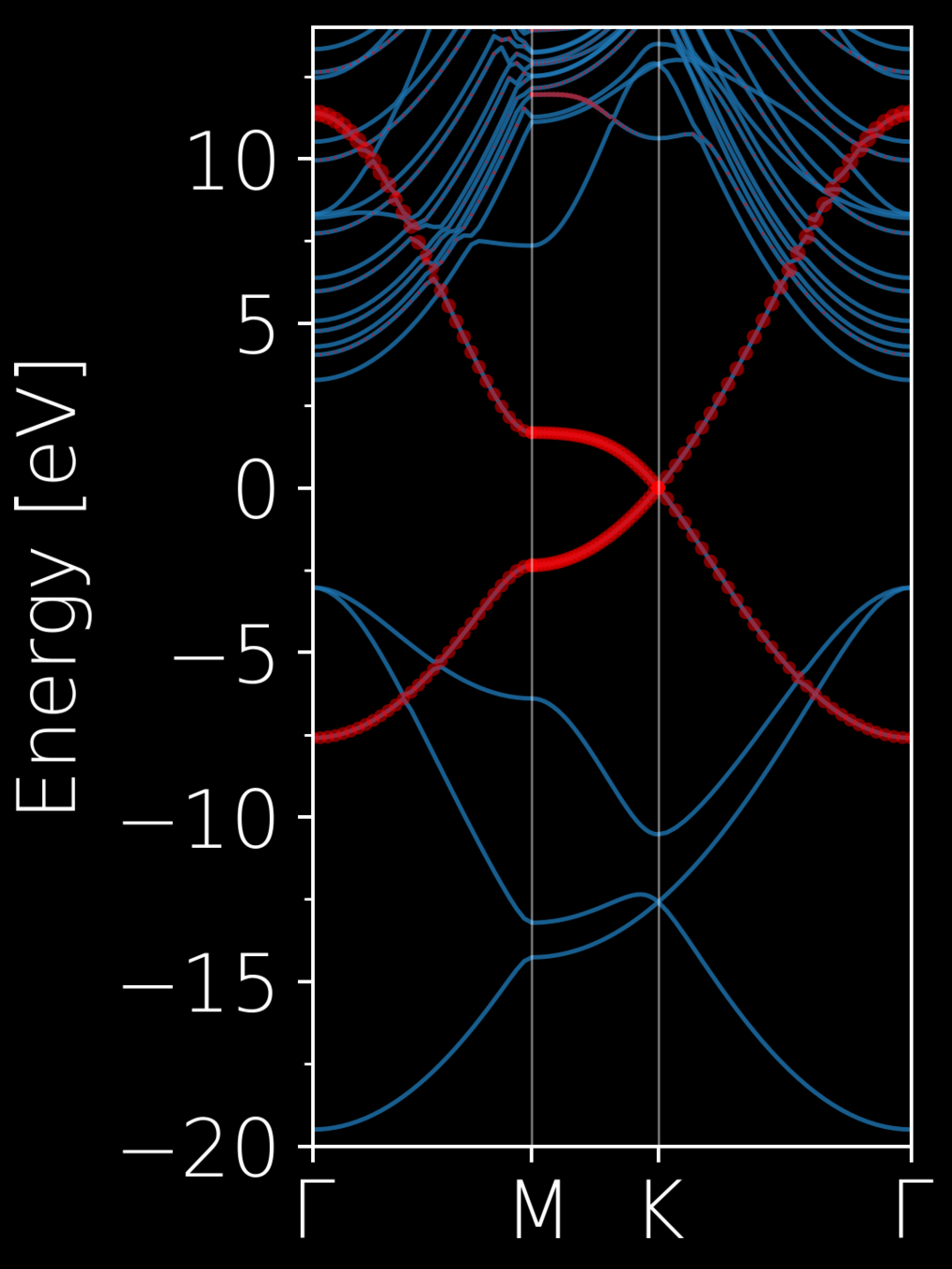
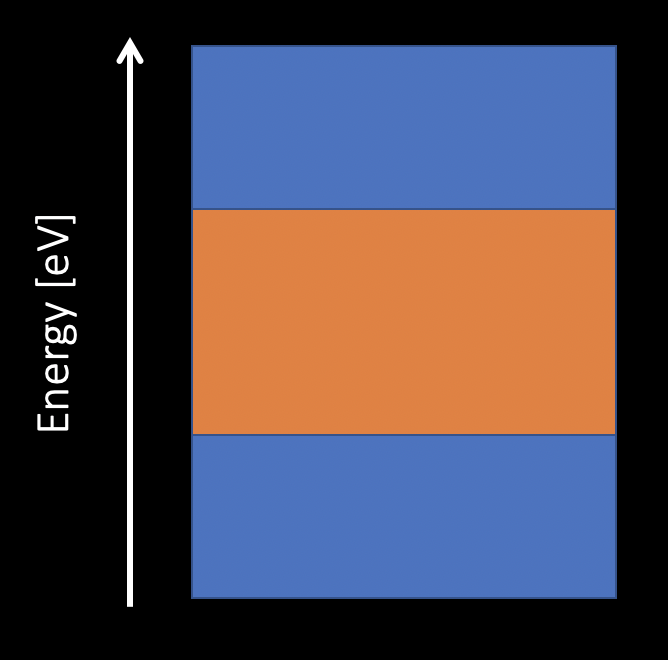
No entanglement
Entanglement
Entanglement
We can use another \( U_{mn}^{\mathrm{(pre)}} \) to do the band selection!
Maximally localized Wannier function!
# band indeices starts from 0
num_bands = 7
num_wann = 4
exclude_bands = 8-16
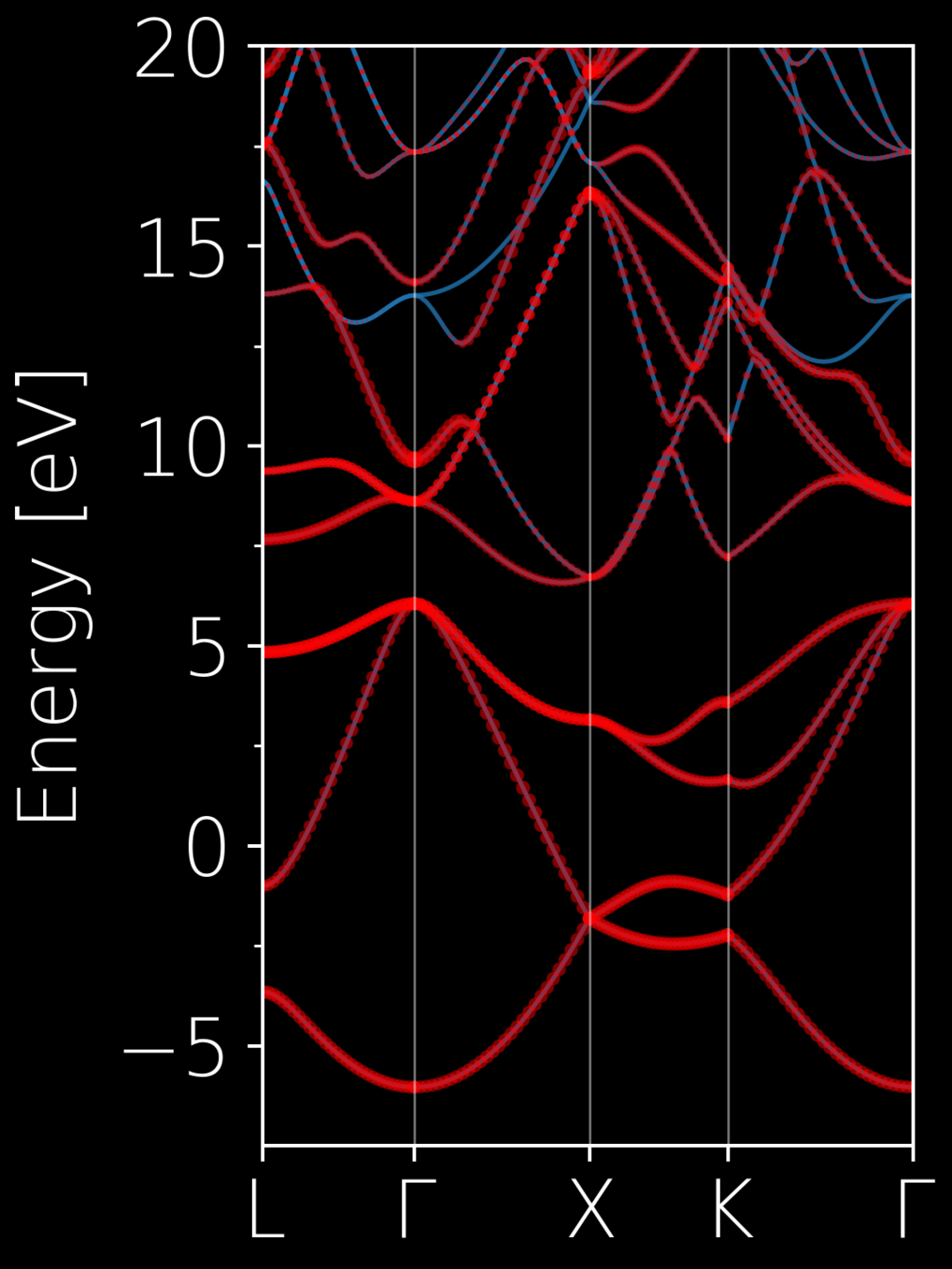
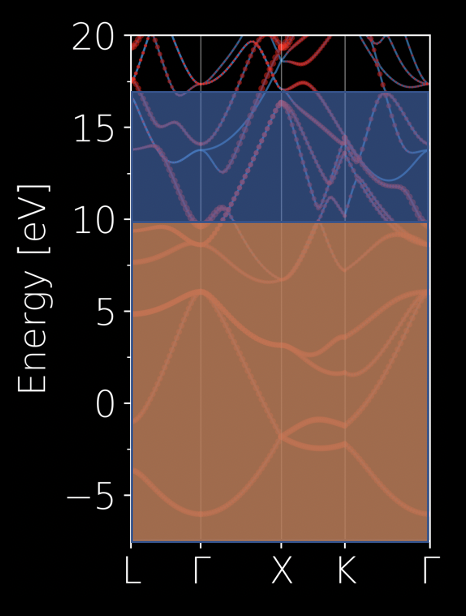
# total window
dis_win_max = 20.0
dis_win_min = -7.5
# frozen window
dis_froz_max = 6.2
dis_froz_min = -7.5
# disentanglement control
#dis_num_iter = 120
#dis_mix_ratio = 1.d0
num_iter = 0
num_print_cycles = 10
# projection section
Begin Projections
c=0.0,0.0,0.0:sp3
End Projections
☄️Some of my work that has something to do with Wannier90:
Find these slides:
Download examples:
By Chengcheng Xiao
A template for ICL+TYC logo imprinted


