SASSIE: Modelling AUC & SAS Data Using Atomistic Simulations
25th July 2017
David Wright & Emre Brookes

Molecular modelling and simulation: What is it?
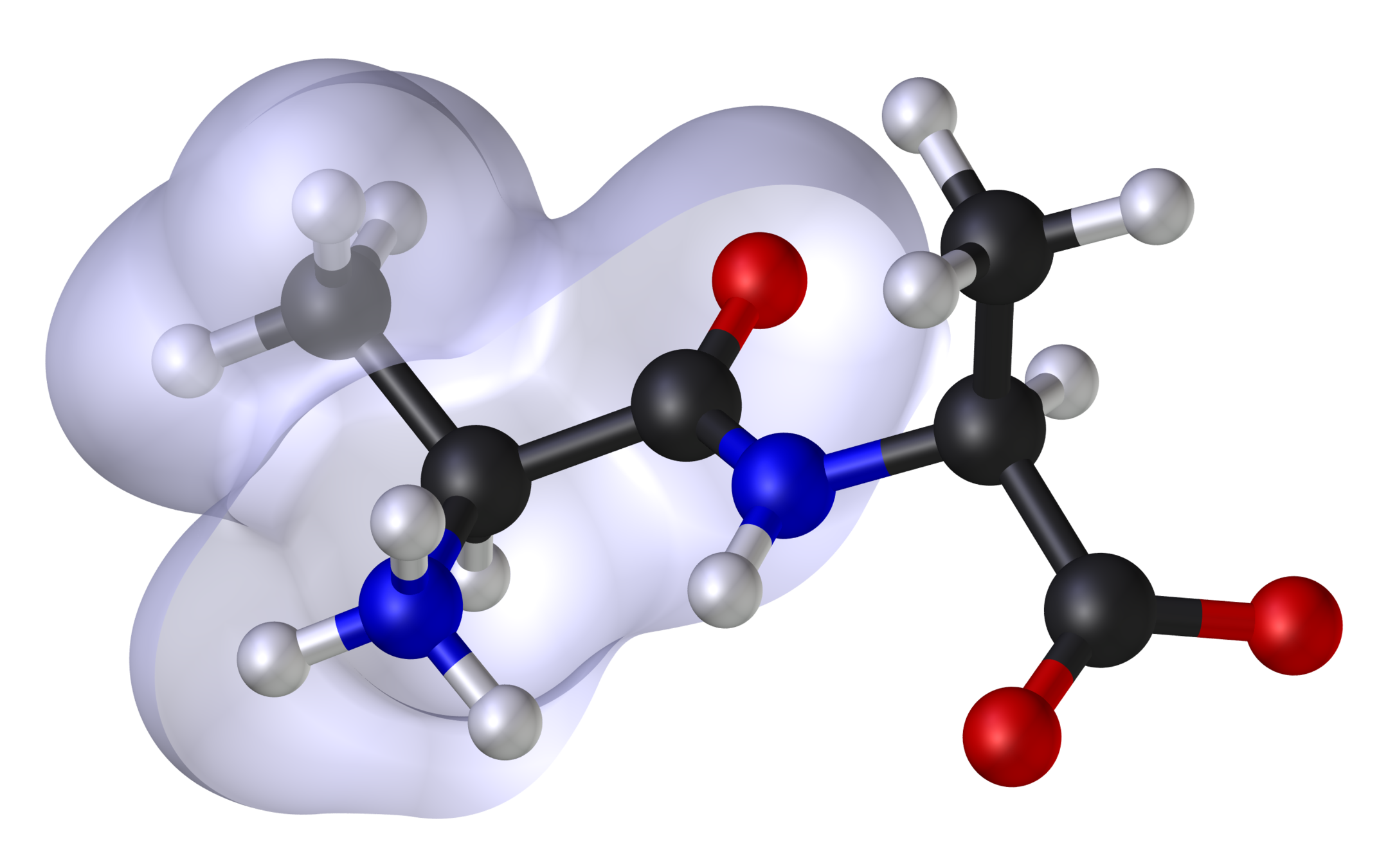
- Theoretical and computational methods used to model or mimic the behaviour or properties of molecules
- Atomistic representation of molecules
- Encode chemistry
- Use classical or quantum mechanics to describe interactions
Molecular modelling and simulation: What is it good for?
- Understanding experimental results
- Combining information from multiple experiments
- Providing atomistic explanations for higher level observations

+
+
What do we need to model AUC & SAS experiments?
- Initial models capturing known chemistry
-
Variety of physically plausible conformations
- Global structure
- Domain level rearrangements
- Atomistic models we can analyse
Molecular Dynamics
Classical (Newtonian) dynamics
F = m a
Forcefield description of interactions
F=−∇ U
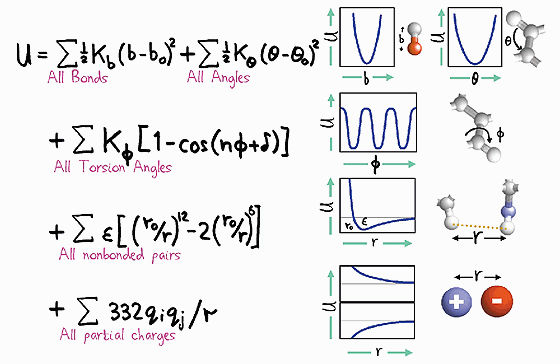
Molecular Dynamics
MD
Experiment
Model system with N particles, solve F = ma until properties do not change (equilibrate) then you “measure” (i.e. average a property) until data converge
Sample in instrument, measure over time ... measure longer until data converge.
Molecular Dynamics
MD and experiment can both suffer from the similar issues
| Experiment | MD |
|---|---|
| Sample not prepared correctly | Incorrect starting model structure |
| Measurement too short | Simulation too short |
| System undergoes irreversible change (aggregation etc.) | Structure trapped in local minima |
| Didn’t quite measure what we thought | Bug in your analysis code |
Molecular Dynamics
Requirements
- Initial structure
- Forcefield
Output
- Trajectory of coordinates
Molecular Dynamics
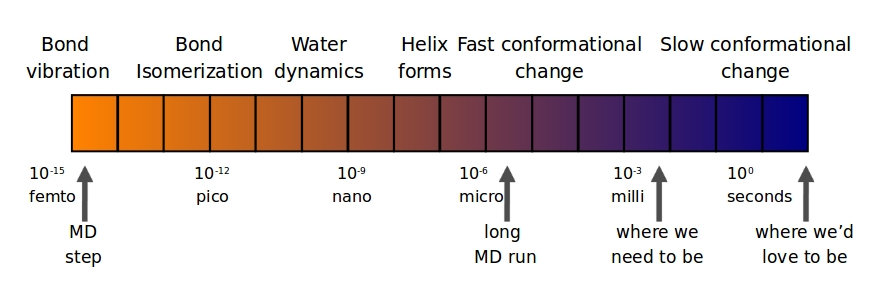
- Time step determined by fastest motion
- Usually X-H bond
- Even fastest supercomputers allow only microsecond simulations in general
Monte Carlo Simulation

- Vary system
- Evaluate energy
-
Keep new structure if
- More energetically favourable or
- With probability related to energy change
- Repeat first step
Dihedral Angle Monte Carlo Simulation
Frenkel and Smit, Understanding Molecular Simulation
Dihedral Angle Monte Carlo Simulation
- MC with all degrees of freedom varied at least as slow as MD
- "Freezing" some degrees of freedom potentially gives a huge speedup
-
Sample only the dihedral potential using the Metropolis criterion
- Dihedral Angle Monte Carlo
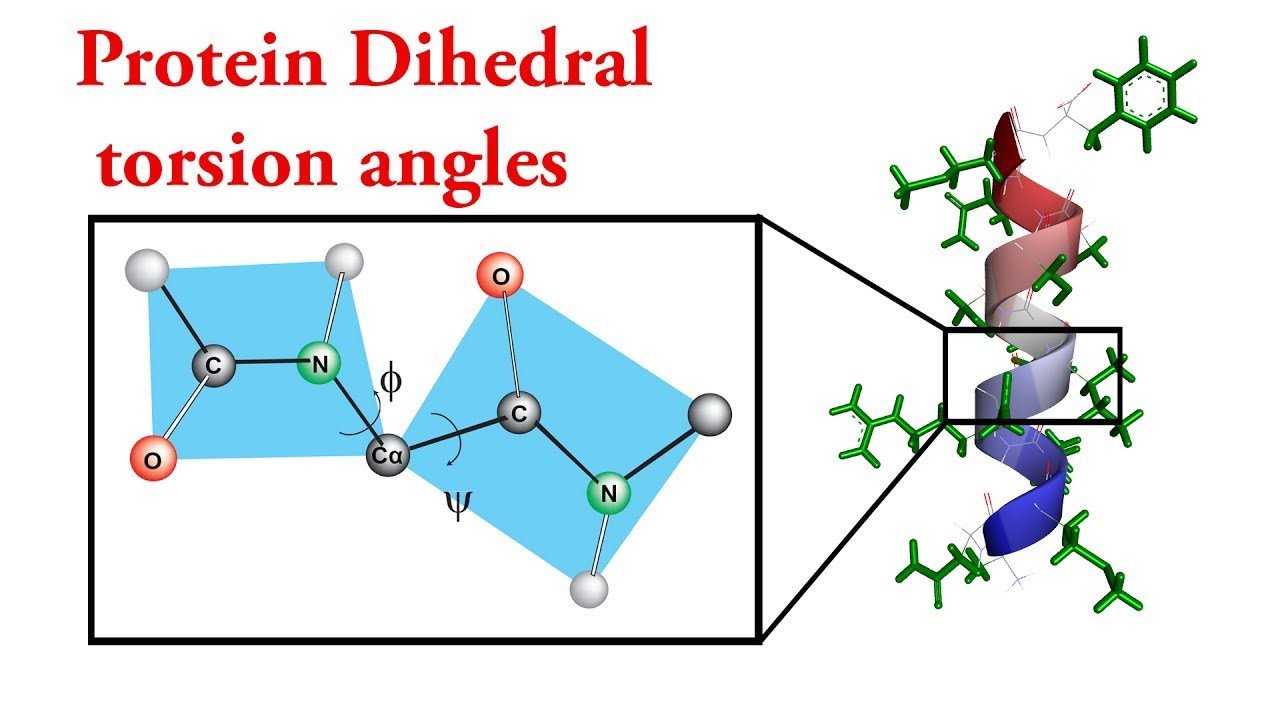
Dihedral Angle Monte Carlo
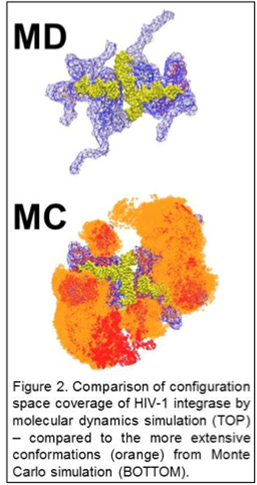
8 cores
13 days
1 core
15 mins
- Rapid generation of ensemble
- Sampling Ramachandran obviously limiting
- Use as hypothesis to test against data
SASSIE
Simple unified interface to tools facilitating:
- Initial model building/preparation
-
Simulation
- Dihedral angle Monte Carlo
- Molecular dynamics
- Torsion angle MD
-
Calculation
- SAS curves
- AUC profiles
- Comparison to experimental data
Further Reading
Full three day training course (today is similar to day 2):
https://sassie-web.chem.utk.edu/training/uk_2017/main.html
MD
A. Leach, Molecular Modelling: Principles and Applications
J. D. Durrant & J. A. McCammon, Molecular dynamics simulations and drug discovery, BMC Biology, 2011, 9:71, DOI: 10.1186/1741-7007-9-71
Dihedral Angle Monte Carlo
J. E. Curtis et al, SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints, Computer Physics Communications, 2012, 183:2, DOI: 10.1016/j.cpc.2011.09.010
Torsion Angle MD
W. Zhang et al, Combined Monte Carlo/torsion-angle molecular dynamics for ensemble modeling of proteins, nucleic acids and carbohydrates, J Mol Graph Model, 2017, 73, DOE: 10.1016/j.jmgm.2017.02.010
AUC 2017 - SASSIE
By David Wright