




















Inês Mendes
Bioinformatics PhD student.
Towards Accreditation in Metagenomics for Clinical Microbiology
Second Thesis Committee Meeting
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
17th of March, 2022
The greatest adventure is what lies ahead.
Today and tomorrow are yet to be said.
The chances, the changes are all yours to make.
The mould of your life is in your hands to break.
– J. R. R. Tolkien, The Hobbit
The global impact of microbial pathogens
A genomic approach to clinical microbiology
The role of bioinformatics
World Health Organisation Global Priority Pathogens list. This catalogue includes, besides Mycobacterium tuberculosis considered the number one global priority, a list of twelve microorganisms grouped under three priority tiers according to their antimicrobial resistance: critical (Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacteriaceae), high (Enterococcus faecium, Helicobacter pylori, Salmonella species, Staphylococcus aureus, Campylobacter species and Neisseria gonorrhoeae), and medium (Streptococcus pneumoniae, Haemophilus influenzae and Shigella species). The major objective was to encourage the prioritisation of funding and incentives, align research and development priorities of public health relevance, and garner global coordination in the fight against antimicrobial-resistant bacteria. Adapted from World Health Organization, 2017.
Bacterial Population Genetics
Pathogenesis and Natural History of Infection
Outbreak Investigation and Control
Surveillance of Infectious Diseases
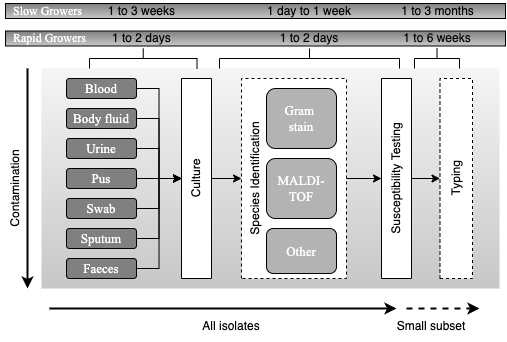
Principles of current processing of bacterial pathogens. Schematic representation of the current workflow for processing samples for bacterial pathogens is presented, with high complexity and a typical timescale of a few weeks to a few months. Samples that are likely to be normally sterile are often cultured on rich medium that will support the growth of any culturable organism. Samples contaminated with colonising flora present a challenge for growing the infecting pathogen. Many types of culture media (referred to as selective media) are used to favour the growth of the suspected pathogen. Once an organism is growing, the likely pathogens are then processed through a complex pathway that has many contingencies to determine species and antimicrobial susceptibility. Broadly, there are two approaches. One approach uses MALDI-TOF for species identification prior to setting up susceptibility testing. The other uses Gram staining followed by biochemical testing to determine species; susceptibility testing is often set up simultaneously with doing biochemical tests. Lastly, depending on the species and perceived likelihood of an outbreak, a small subset of isolates may be chosen for further investigation using a wide range of typing tests. Adapted from Didelot et al., 2012
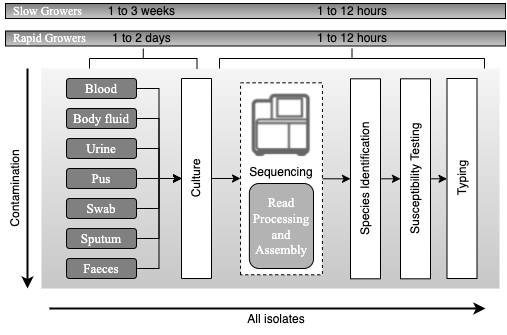
Principles of current processing of bacterial pathogens based on whole-genome sequencing. Schematic representation of the workflow for processing samples for bacterial pathogens after the adoption of whole-genome sequencing, with an expected timescale that could fit within a single day. The culture steps would be the same as currently used in a routine microbiology laboratory. Once a likely pathogen is ready for sequencing, DNA will be extracted, taking as little as 2 hours to prepare the DNA for sequencing. After sequencing, the main processes for yielding information will be computational. Automated sequence assembly algorithms are necessary for processing the raw sequence data, from which species, relationship to other isolates of the same species, antimicrobial resistance profile and virulence gene content can be assessed. All the results will also be used for outbreak detection and infectious diseases surveillance Adapted from Didelot et al., 2012
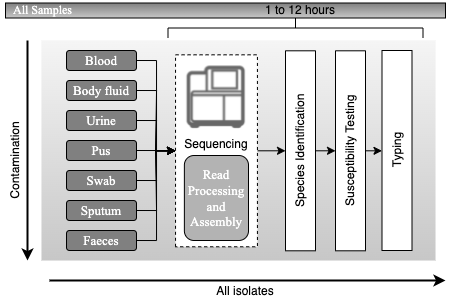
Hypothetical workflow based on metagenomic sequencing. Schematic representation of the hypothetical workflow for the direct processing of samples from suspected sources of pathogens after adoption of metagenomic sequencing, with an expected timescale that could fit within a single day. Adapted from Didelot et al., 2012
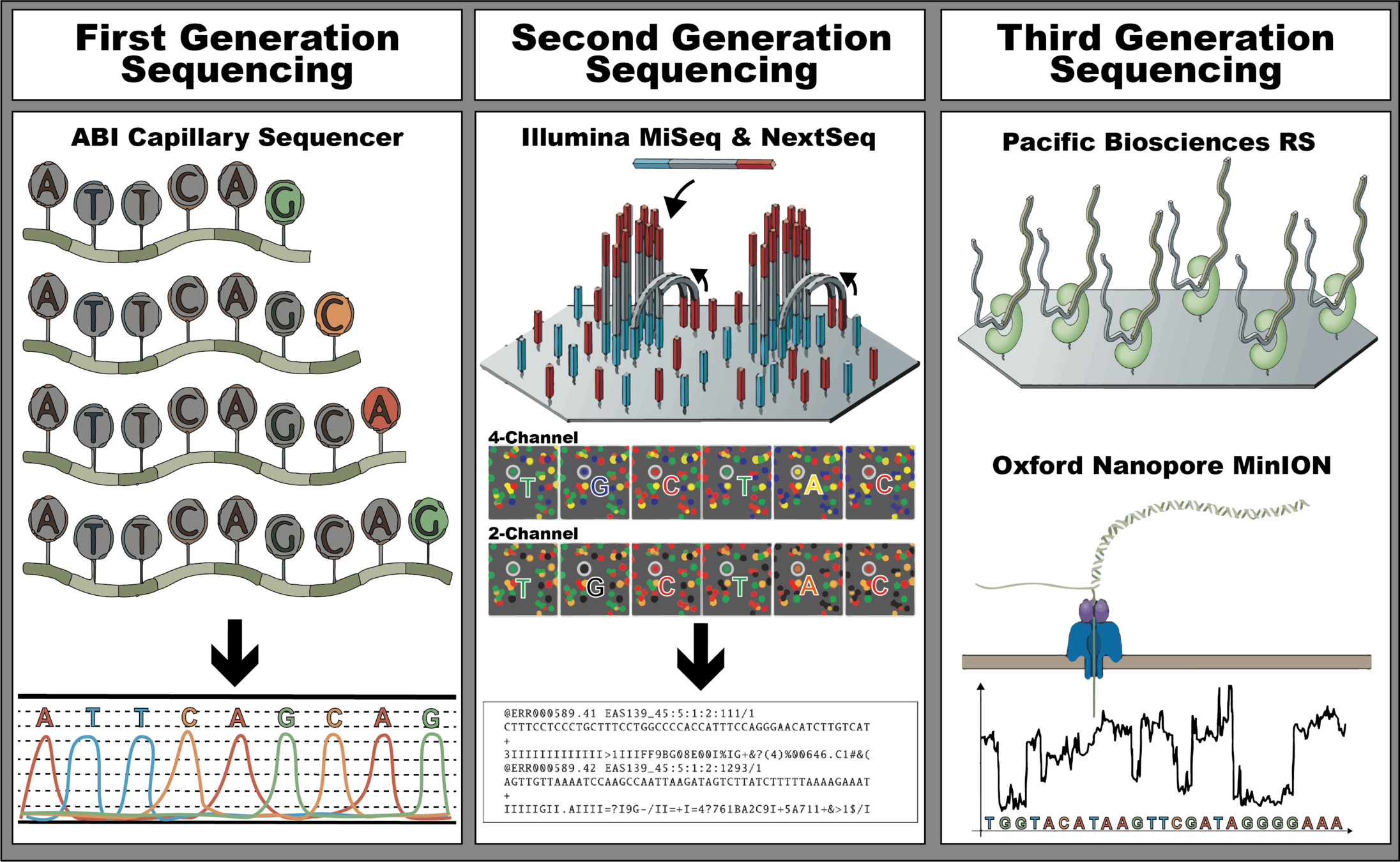
The three revolutions in sequencing technology that have transformed the landscape of bacterial genome sequencing. The first-generation, also known as Sanger sequencers, is represented by the ABI Capillary Sequencer (Applied Biosystems). The second-generation, also known as high-throughput sequencers, is represented by the MiSeq, a 4-channel sequencer, and the NextSeq, a 2-channel sequencer (Illumina), both sequencing by synthesis instruments. These instruments allow the sequencing of both ends of the DNA fragment. Lastly, the third-generation, also known as long-read sequencers, is represented by Pacific Bioscience BS sequencer and Oxford Nanopore MinION sequencer. Adapted from Hagemann, 2015; Nicholas J. Loman
and Pallen, 2015; Goodwin et al., 2016; Wang et al., 2021; Metzker, 2010; Xu et al., 2020.
Read mapping
Read mapping software
reads
reference genome
Gene-by-Gene
de novo assembly software
reads
contigs
annotation/ comparison
11 CLINICAL METAGENOMIC SAMPLES
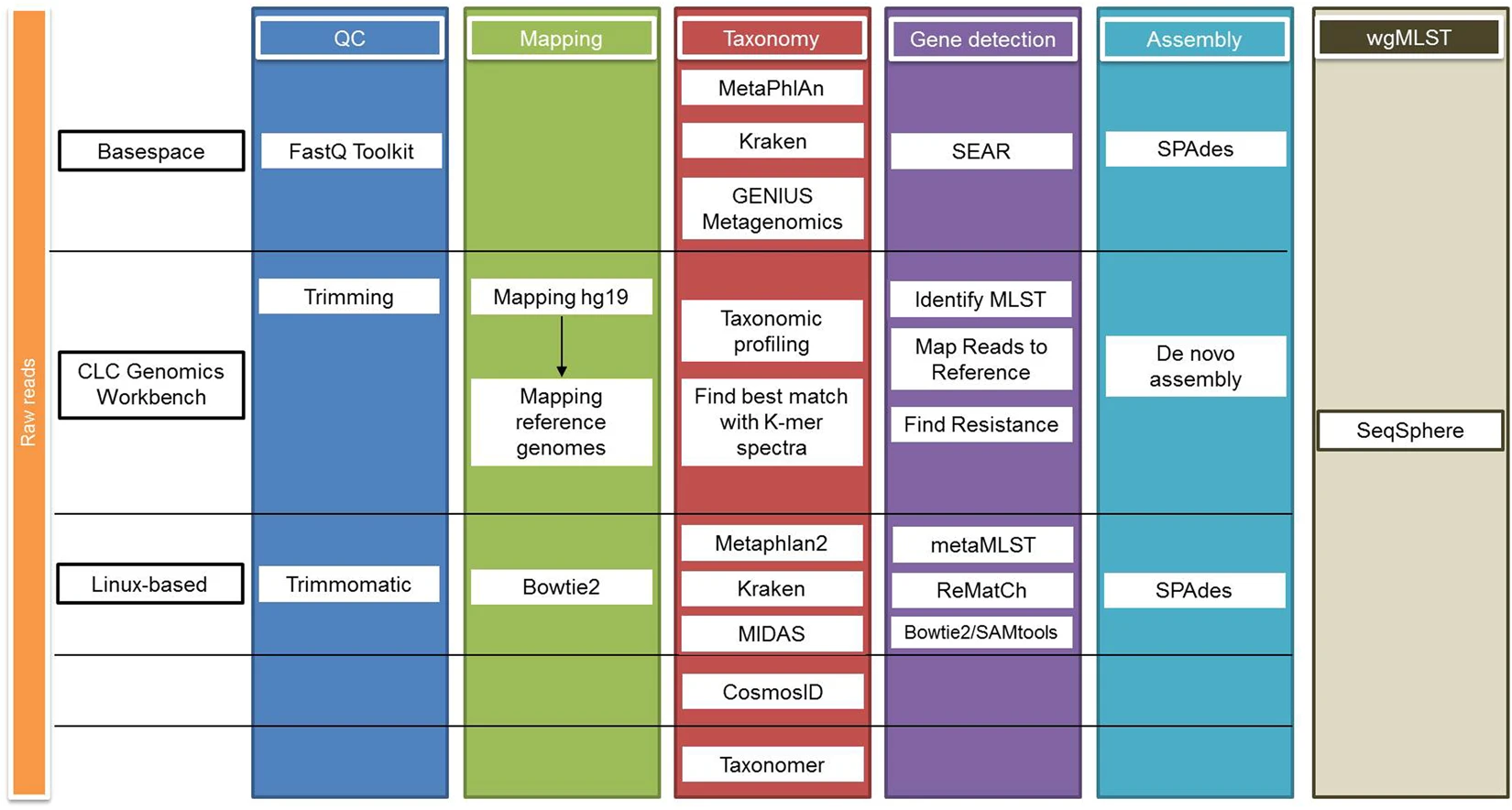
Scheme of the bioinformatic analysis of the metagenomics samples. In order to evaluate and compare the accuracy and reliability of the bioinformatics analyses in providing the closest results to culture and WGS of any cultured isolates, three different pipelines (two commercially and one freely available) were used (Fig. 1). Different tools to perform raw read quality control, filtering and trimming were used and reads were mapped against the human genome (hg19) before performing taxonomic classification. Reads mapping to hg19 were removed from the analysis to increase the efficiency of the bioinformatics tools. Typing (MLST), phylogenetic analysis, plasmid analysis, detection of antimicrobial resistance and virulence genes was performed. To determine the appropriateness of shotgun metagenomics as a predictor of the WGS (chromosome and plasmids), Shotgun metagenomics results obtained were compared with the results of WGS of any bacterial isolates obtained from culturing the sample. Source: Couto et al, 2018.
Precision, Sensibility & Performance
(Culture + Maldi-TOF)
Highlights the potential and the limitations of shotgun metagenomics as a diagnostic tool
Results are highly dependent on the tools, and specially database, chosen for the analysis
1 SURVAILANCE METAGENOMIC SAMPLE
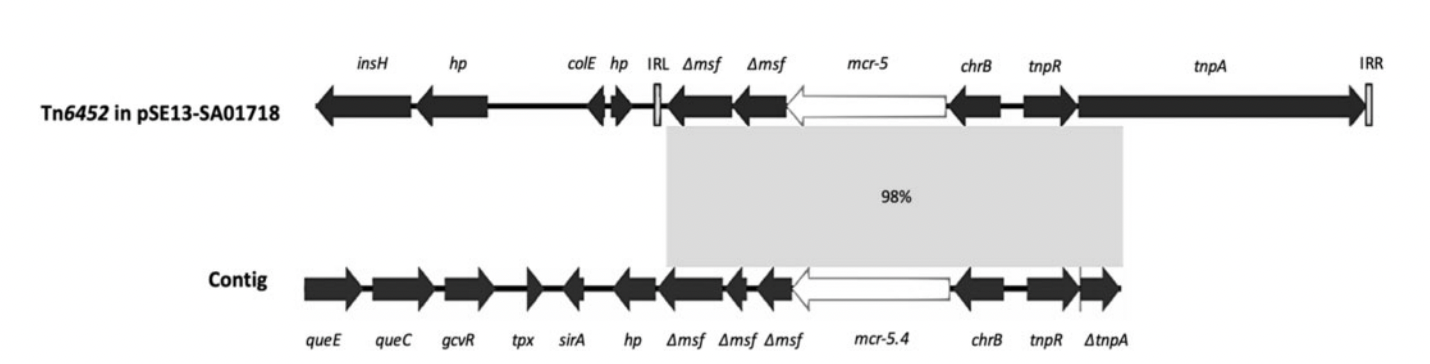
Comparative analysis of the genetic environment of mcr-5 between the reference plasmid pSE13-SA01718 (accession no. KY807921.1) and the annotated hybrid metagenome contig (accession no. MK965519). The contig carrying the mcr-5.4 gene consists of the following putative gene products: 7-carboxy-7-deazaguanine synthase (queE), 7-cyano-7-deazaguanine synthase (queC), glycine cleavage system transcriptional antiactivator GcvR (gcvR), thiol peroxidase (tpx), sulphurtransferase TusA family protein (sirA), hypothetical protein (hp), truncated MFS-type transporter (Dmsf), lipid A phosphoethanolamine transferase (mcr-5.4), ChrB domain protein (chrB), transposon resolvase (tnpR) and truncated transposon transposase (DtnpA). Areas with 98% identity between sequences are represented in light grey. Arrows indicate the position and direction of the genes. The transposon Tn6452 sequence in the reference plasmid pSE13-SA01718 is bounded by inverted repeats: IRL and IRR.Source: Fleres et al, 2019.
short reads
long reads
de novo hybrid assembly
annotation/ comparison
Even when hybrid assembly is employed, complete genomic sequences, particularly chimeric ones such as plasmids, are not fully recovered.
Results are highly dependent on the tools, and specially database, chosen for the analysis
Dengue virus genomics
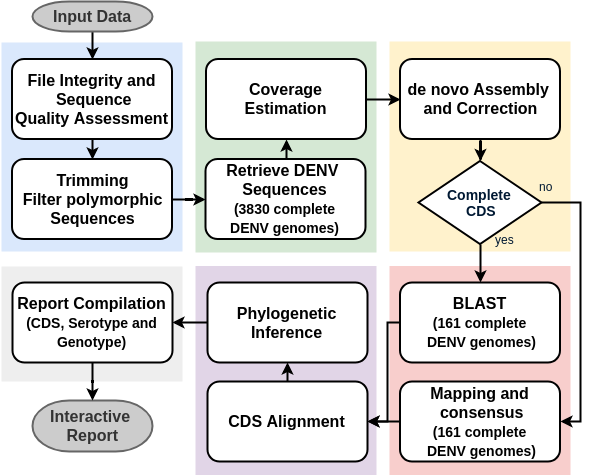
The DEN-IM workflow
https://doi.org/10.1371/journal.pntd.0001876.g002
https://doi.org/10.1371/journal.pntd.0000757
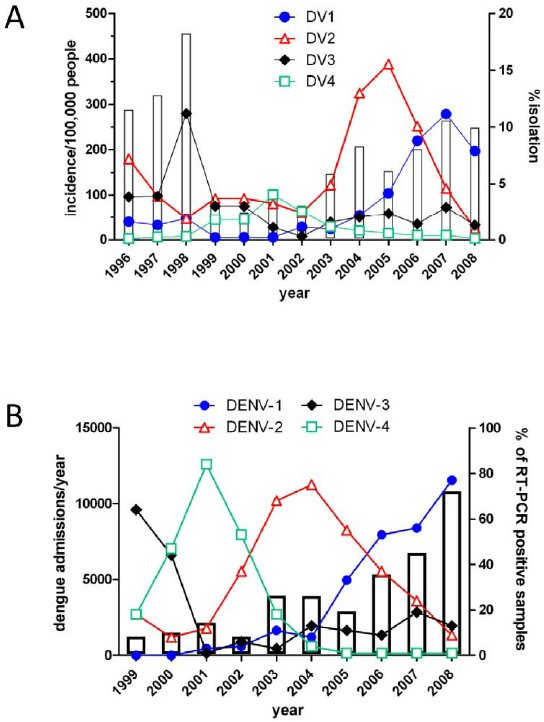
Thailand
Viet Nam
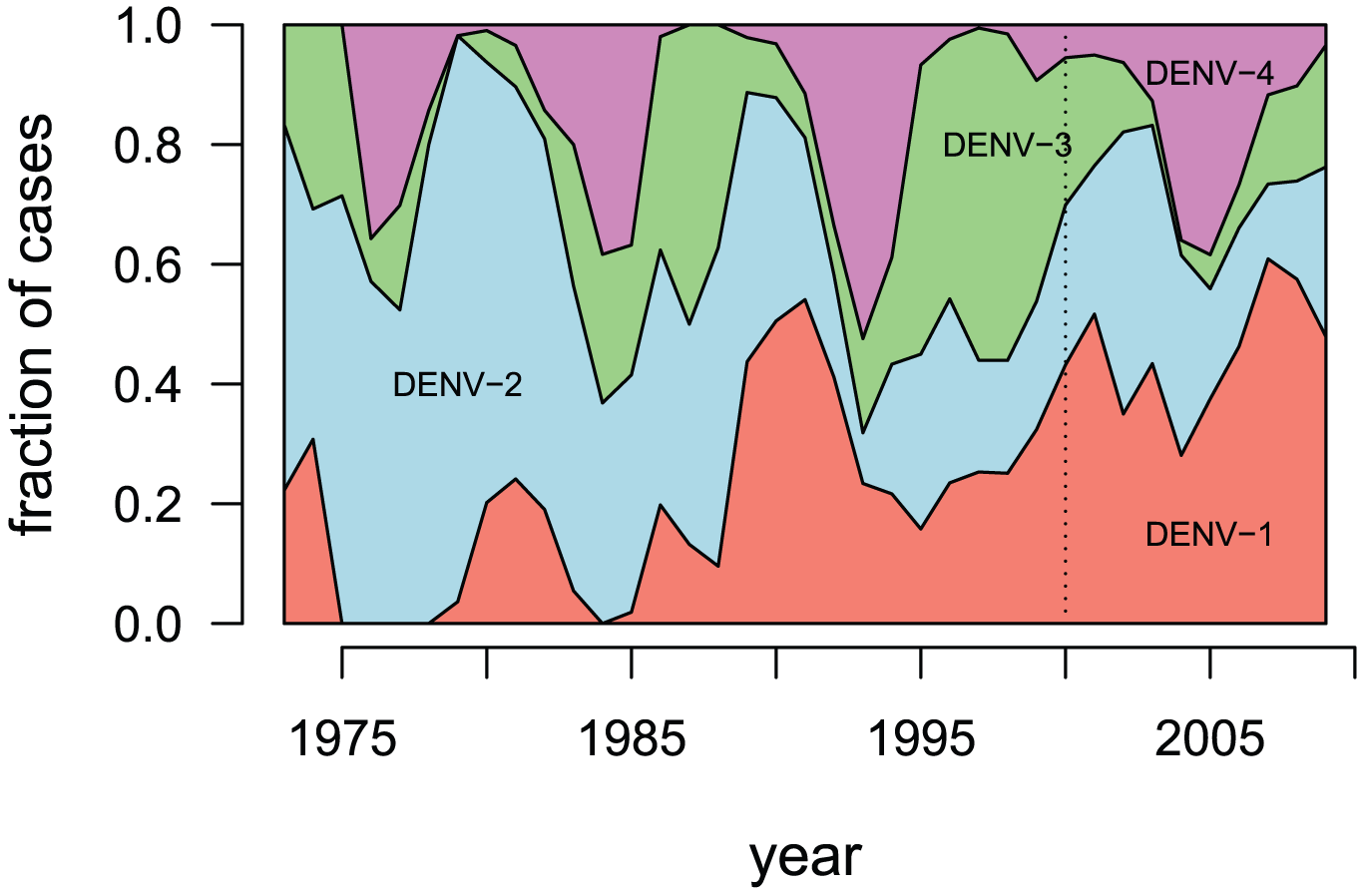
Sequential infection increases the risk of a severe form of the infection - dengue hemorrhagic fever.
Requirements
A solution
DENV Identification
In Silico Typing:
Leveraging the use of container software with workflow managers enables reproducible and collaborative research
Stand-alone HTML reports allow for interactive report exploration across domains
The LMAS workflow
https://github.com/cimendes/LMAS
https://lmas.readthedocs.io/
Computational costs and performance on organisms of interest must be taken into consideration when choosing the most appropriate assembly software
Some assemblers still in use, such as ABySS, BCALM2, MetaHipmer2, minia and VelvetOptimiser, perform relatively poorly and should be used with caution.
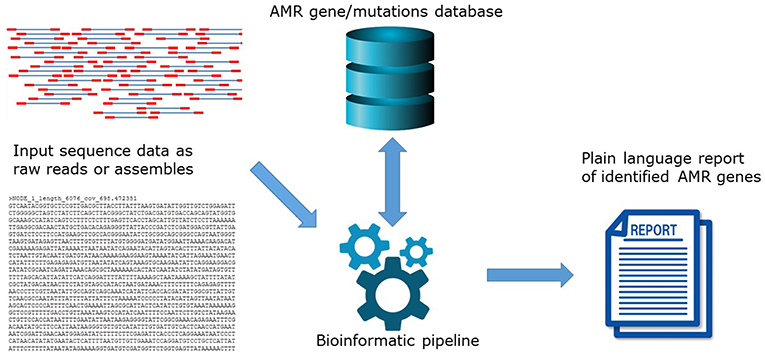
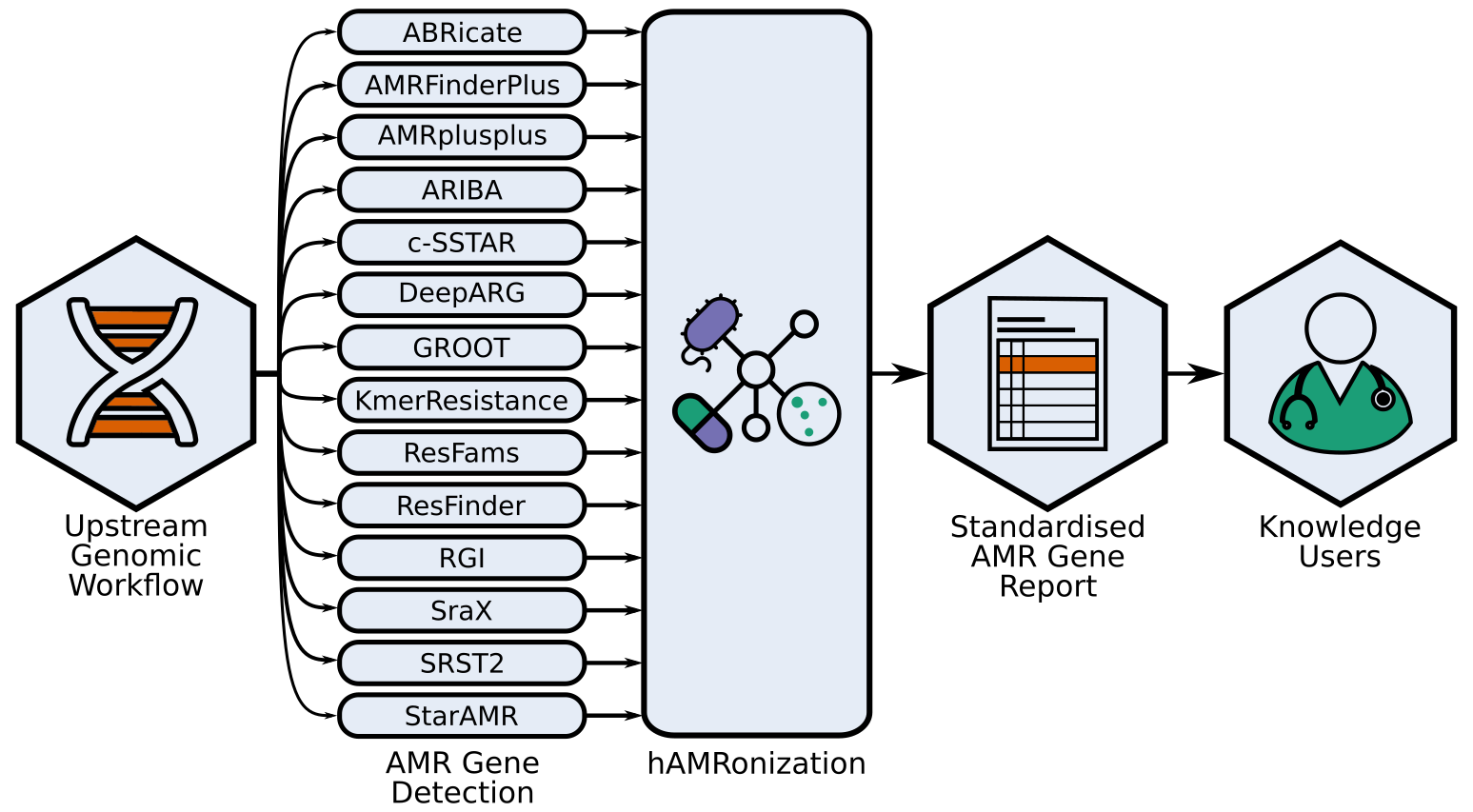
The hAMRonization package
https://doi.org/10.3389/fpubh.2019.00242
The lack of standardization greatly hinders the comparison of results across sectors. The myriad of options available for this purpose highlights a grave interoperability problem.
This allows allowing not only the comparison of tools and databases but the validation of results through multiple detection algorithms.
The SARS-CoV-2 Data Specification
The FAIR Data Principles: Findable, Accessible, Interoperable, and Reusable
This allows allowing not only the comparison of tools and databases but the validation of results through multiple detection algorithms.
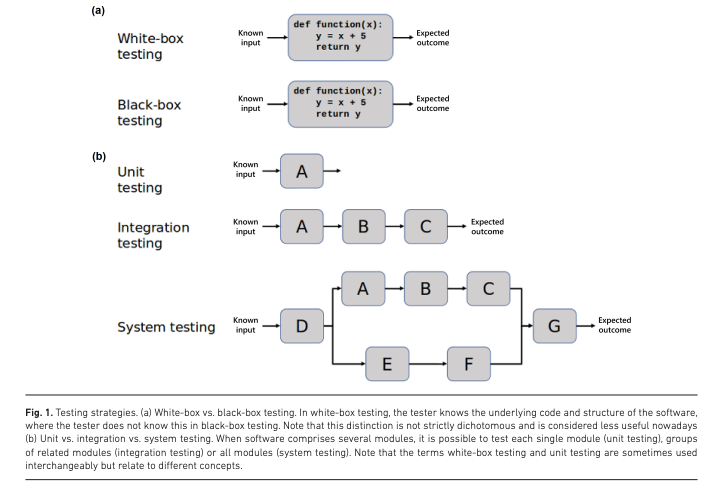
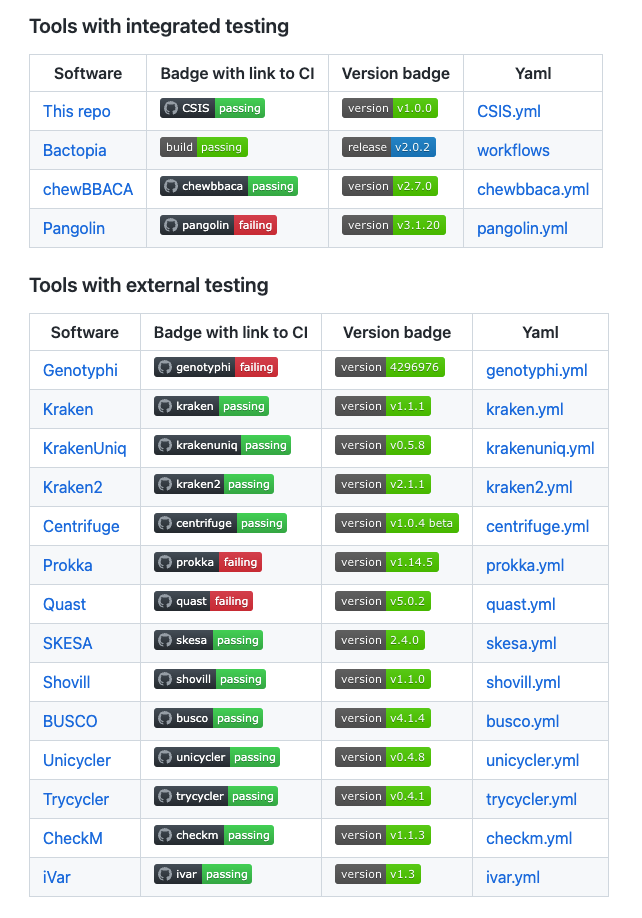
A call to action for software testing
The CSIS repository aims to promote the uptake of testing practices and engage the community in its adoption for public health.
This repository is an open-source project that gathers guidance, guidelines and examples for software testing for microbial bioinformatics researchers.
A proof of concept that the adoption of new standards can be crowdsourced.
The use of software testing ensures not only that the tool is working as expected, but how it can be leveraged to be used as proxy for workability
Crowdsourcing proved to be a reliable strategy for the adoption of new standards in the microbial bioinformatics community
Lack of reproducibility and standardisation is a major hinder in metagenomics for clinical microbiology.
Even with the use of long-reads complex genomic regions, such as chimeric plasmids, are a challenge to retrieve.
Leveraging the use of container software with workflow managers represents the current best standard for reproducible research.
Intuitive and responsive reports enable collaborative research and empowers users across domains.
Benchmark of basal tools, such as de novo assemblers, highlights the need for proper software assessment.
Standard specifications, such as for AMR or SARS-COV-2, are required for the comparison of results across stakeholders in different domains.
Crowdsourcing for better standards represents a viable way to adopt better practices for the use of metagenomics in clinical microbiolgy.
Thank you for
your attention
SFRH/BD/129483/2017
By Inês Mendes
CAML PhD Program Thesis Committee - 17 March 2022




