

















Inês Mendes
Bioinformatics PhD student.
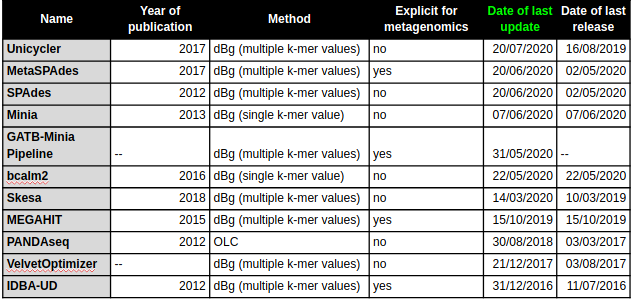
Benchmarking of de novo (meta)genomic assembly software
Computational Biology and Bioinformatics Day
October 21, 2020
@ines_cim
cimendes
Inês Mendes
M Ramirez Lab
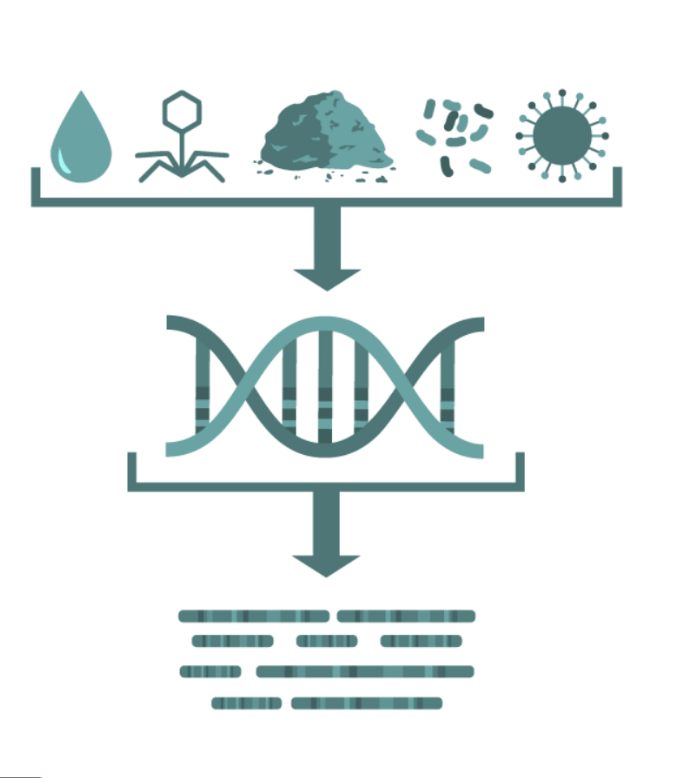
Random "shotgun" sequencing of microbial DNA, without selecting a particular gene.
Promising methodology for obtaining fast results for the identification of pathogens and their virulence and antimicrobial resistance properties without the need for culture.
The assembly methods provide longer sequences that are more informative than shorter sequencing data and can provide a more complete picture of the microbial community in a given sample.
Reference Dataset (Complete Bacterial Genomes)
In silico mock sample (even)
In silico mock sample (log)
Zymos standard (even)
Zymos standard (log)
3.7 M read pairs
8.8 M read pairs
47.8 M read pairs
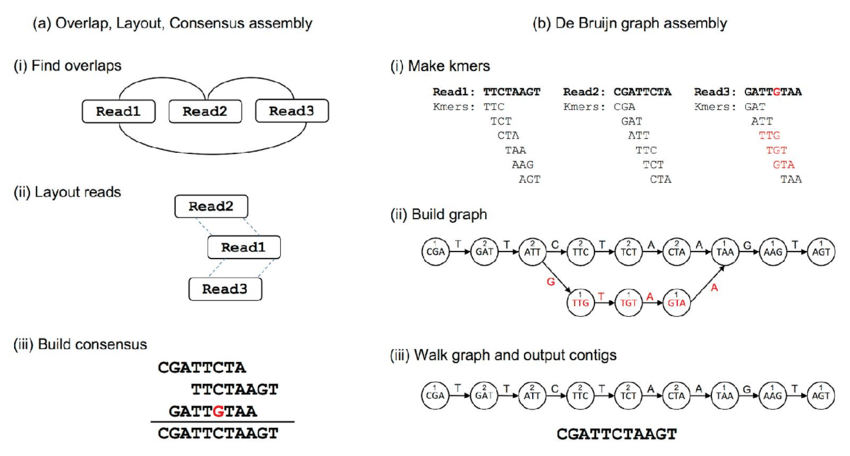
Assembly Workflow
Assembly Quality Assessment
Reference Dataset (Triple)
Assembly file (fasta)
Filter min contig size (1000 bp)
Mapping with Minimpa2
Read Data
PAF file (tab)
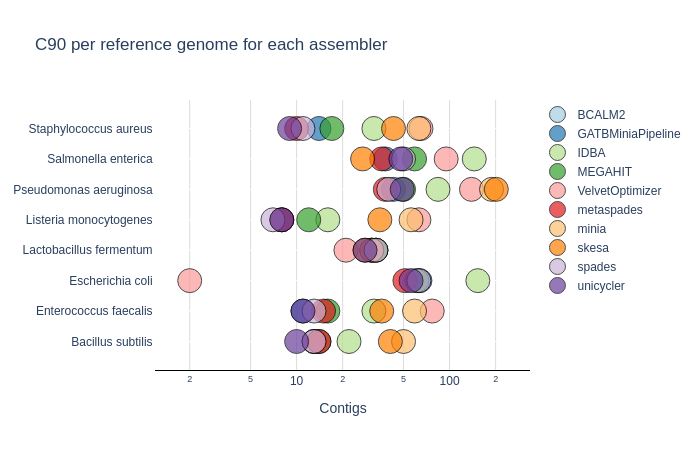
C90 & C95
Number of contigs to cover at least 90% and 95% of the reference genome, respectively.
Contig Phread Quality Score
Contiguity
Longest percentage of the reference sequence assembled in a single contig.
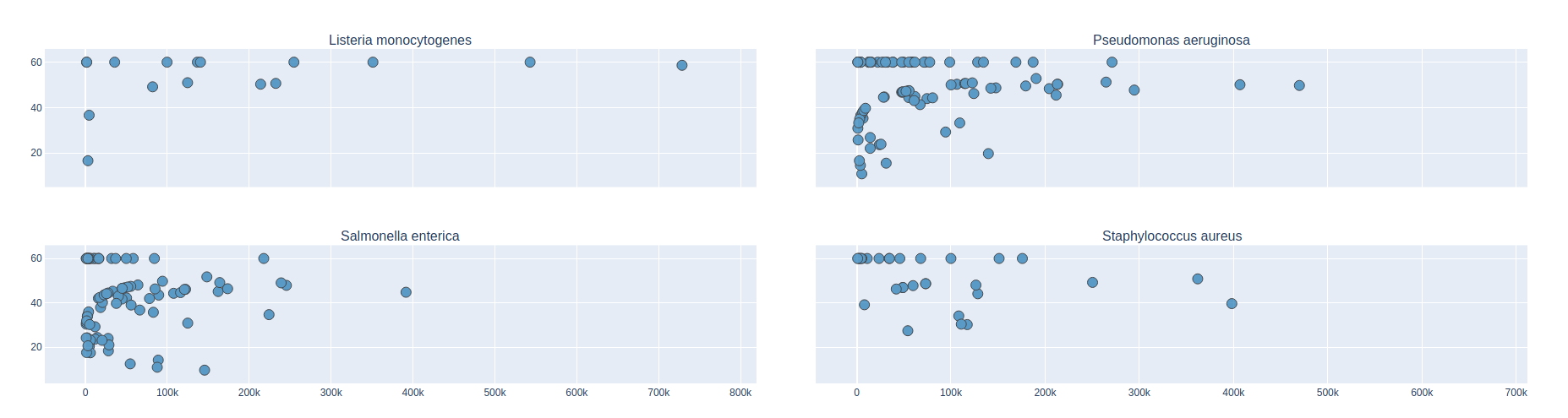
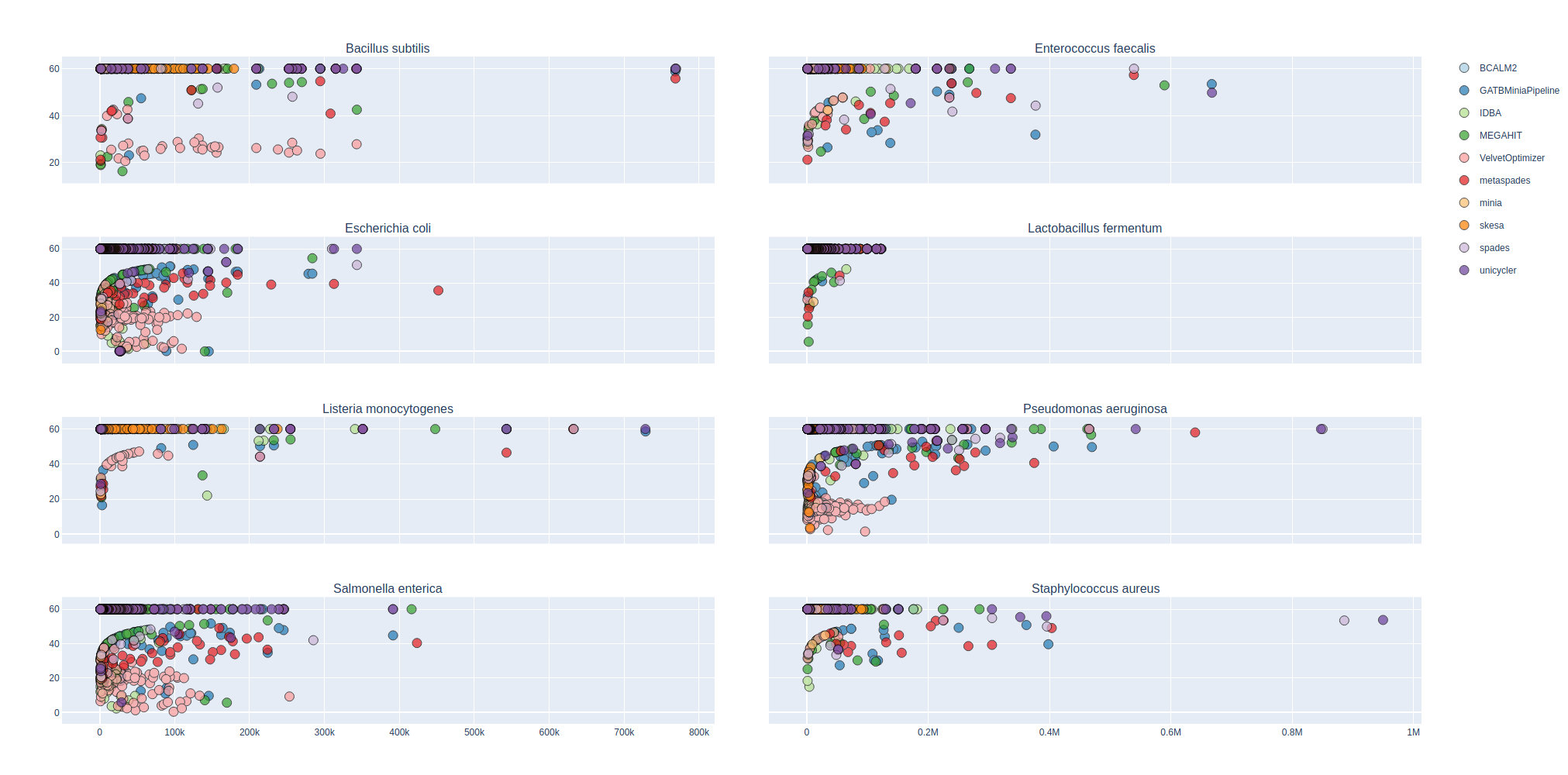
Contig Phred Quality Score for GATBMiniaPipeline's Pseudomonas aerugiona assemby
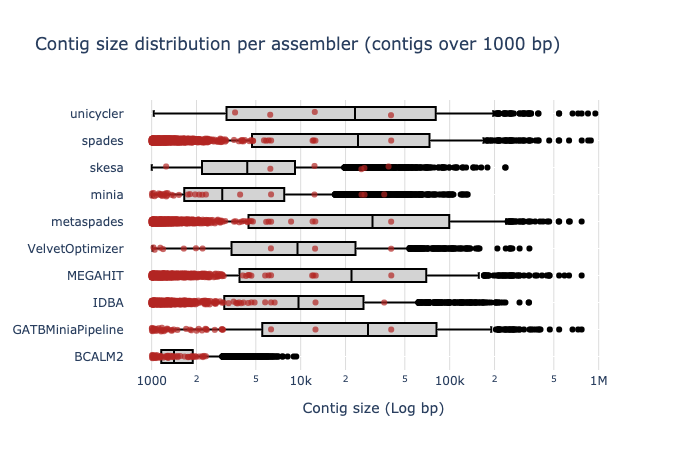
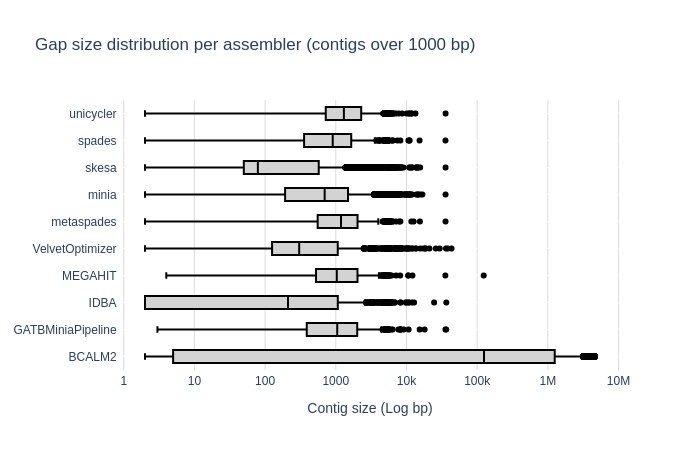
Contig Size
Phred Score
Contig Phred Quality Score per Reference for each Assembler
Special thanks to Pedro Vila-Cerqueira, Rafael Maria Mamede and Mário Ramirez.
FCT PhD Grant SFRH/BD/129483/2017
By Inês Mendes
Slide deck for CBBD's 3 minute presentation




