













Inês Mendes
Bioinformatics PhD student.
Dengue Virus Typing from Shotgun and Targeted Metagenomics
Computational Biology and Bioinformatics Seminars
Inês Mendes
@ines_cim
cimendes
The dengue virus
| Introduction
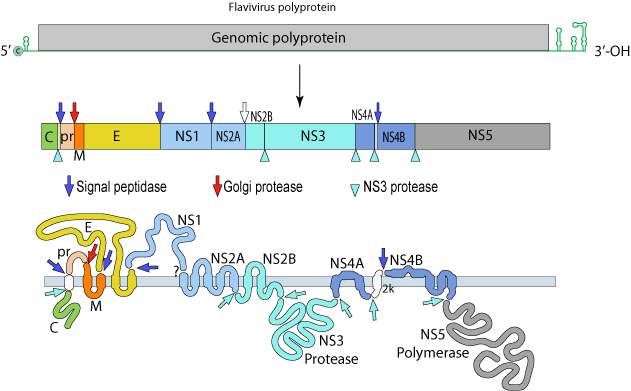
DENV: (+)ssRNA (~11Kb; 1 ORF)
The single polyprotein encodes:
Structural Proteins:
C – capsid
prM – pre-membrane
M - membrane
E - envelope
Non-Structural Proteins:
NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5
| Introduction
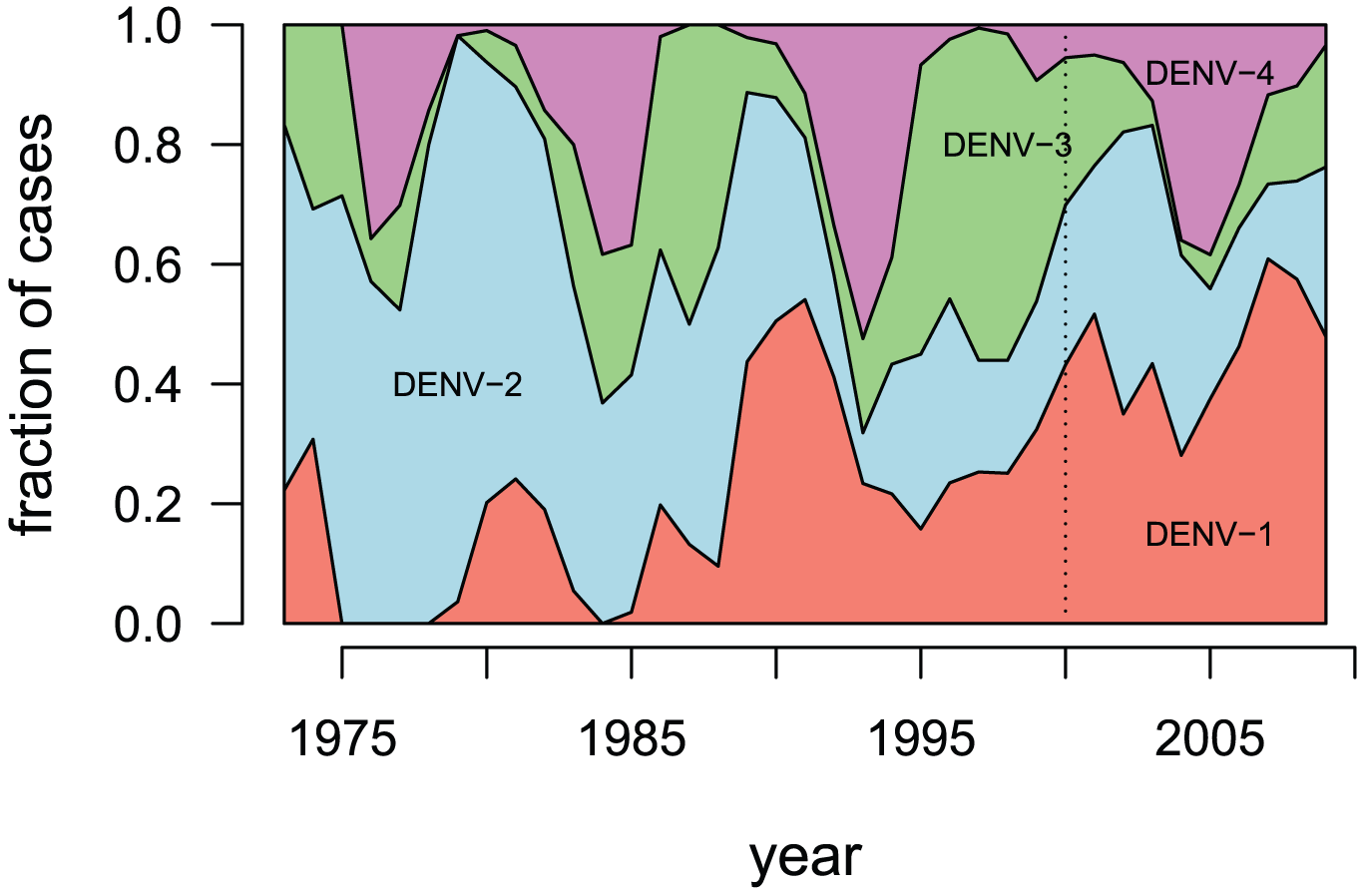
Can be classified into 4 serotypes:
The dengue virus
| Introduction
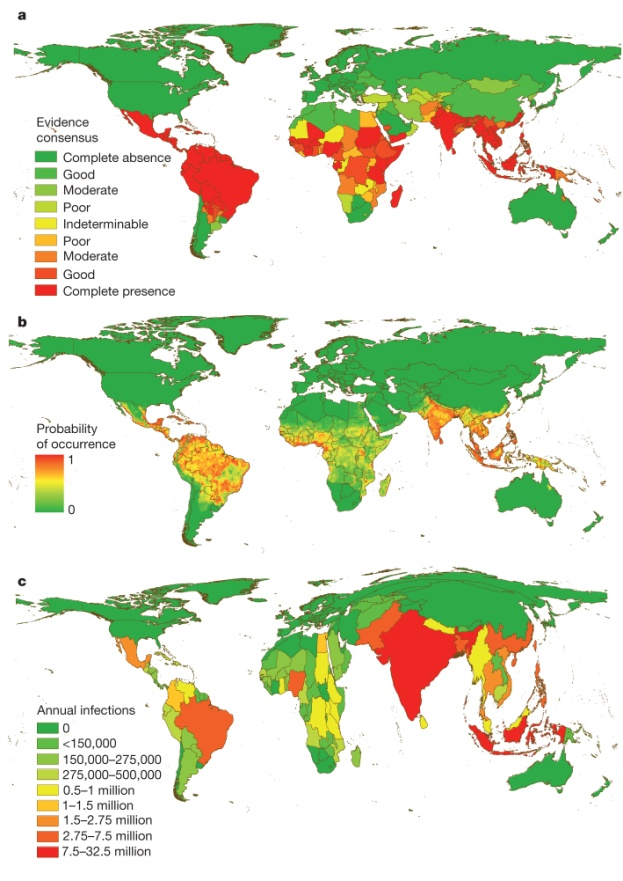
The control of dissemination and characterization of DENV through the use of HTS presents the most promising strategy to understand transmission and disease in populations where it’s infection is endemic.
The dengue virus
| Introduction
The dengue virus
DEN-IM
| The workflow
What is it?
A ready-to-use one-stop and reproducible bioinformatic analysis workflow for the processing and phylogenetic analysis of DENV using paired-end raw sequencing data.
What for?
Empower the use of HTS to monitor the dissemination of the disease directly from patient samples.
DEN-IM
| The workflow
What do I need?
Git, Nextflow (java) and a container engine (Docker, singularity, shifter...).
apt-get install gitcurl -s https://get.nextflow.io | bash
apt-install docker-ceClone (or run remotely)
git clone https://github.com/B-UMMI/DEN-IM.gitDEN-IM
| The workflow
nextflow run DEN-IM.nf --help -profile dockerN E X T F L O W ~ version 0.32.0
Launching `DEN-IM.nf` [nice_monod] - revision: 97bee38c5e
============================================================
D E N - I M
============================================================
Usage:
nextflow run DEN-IM.nf
--fastq Path expression to paired-end fastq files. (default: fastq/*_{1,2}.*)
--genomeSize Genome size estimate for the samples in Mb. It is used to estimate the coverage and other assembly parameters andchecks (integrity_coverage;check_coverage;assembly_mapping)
--minCoverage Minimum coverage for a sample to proceed. By default it's setto 0 to allow any coverage (integrity_coverage;check_coverage)
--adapters Path to adapters files, if any. (fastqc_trimmomatic)
--trimSlidingWindow Perform sliding window trimming, cutting once the average quality within the window falls below a threshold. (fastqc_trimmomatic)
--trimLeading Cut bases off the start of a read, if below a threshold quality. (fastqc_trimmomatic)
--trimTrailing Cut bases of the end of a read, if below a threshold quality. (fastqc_trimmomatic)
--trimMinLength Drop the read if it is below a specified length. (fastqc_trimmomatic)
--clearInput Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (fastqc_trimmomatic;filter_poly;bowtie;retrieve_mapped;viral_assembly;pilon)
--pattern Pattern to filter the reads. Please separate parametervalues with a space and separate new parameter sets with semicolon (;). Parameters are defined by two values: the pattern (any combination of the letters ATCGN), and the number of repeats or percentage of occurence. (filter_poly)
--reference Specifies the reference genome to be provided to bowtie2-build. (bowtie)
--index Specifies the reference indexes to be provided to bowtie2. (bowtie)
--minimumContigSize Expected genome size in bases (viral_assembly)
--spadesMinCoverage The minimum number of reads to consider an edge in the de Bruijn graph during the assembly (viral_assembly)
--spadesMinKmerCoverage Minimum contigs K-mer coverage. After assembly only keep contigs with reported k-mer coverage equal or above this value (viral_assembly)
--spadesKmers If 'auto' the SPAdes k-mer lengths will be determined from the maximum read length of each assembly. If 'default', SPAdes will use the default k-mer lengths. (viral_assembly)
--megahitKmers If 'auto' the megahit k-mer lengths will be determined from the maximum read length of each assembly. If 'default', megahit will use the default k-mer lengths. (default: auto) (viral_assembly)
--minAssemblyCoverage In auto, the default minimum coverage for each assembled contig is 1/3 of the assembly mean coverage or 10x, if the mean coverage is below 10x (assembly_mapping)
--AMaxContigs A warning is issued if the number of contigs is overthis threshold. (assembly_mapping)
--splitSize Minimum contig size (split_assembly)
--typingReference Typing database. (dengue_typing)
--includeNCBI Include NCBI DENV references in alignment. (mafft))
--getGenome Retrieves the sequence of the closest reference. (dengue_typing)
--substitutionModel Substitution model. Option: GTRCAT, GTRCATI, ASC_GTRCAT, GTRGAMMA, ASC_GTRGAMMA etc (raxml)
--seedNumber Specify an integer number (random seed) and turn on rapid bootstrapping (raxml)
--bootstrap Specify the number of alternative runs on distinct starting trees (raxml)
--simpleLabel Simplify the labels in the newick tree (for interactive report only) (raxml)
DEN-IM
| The workflow
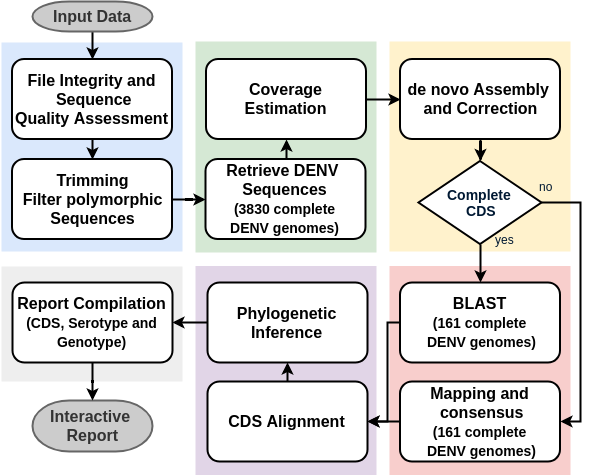
Quality Control:
DENV Sequence Retrieval:
Bowtie2, Samtools
Assembly:
Spades, MEGAHIT
In Silico Typing:
Seq_typing (assembly); Seq_Typing reads (consensus)
Alignment:
MAFFT
Phylogenetic Inference:
RaxML
DEN-IM
| The workflow
DENV Sequence Retrieval:
3830 complete DENV genomes from the NIAID Virus Pathogen Database and Analysis Resource (ViPR)
complete genome sequence
human host (exception of DENV-1 III, monkey)
collection year (1950-2018)
In Silico Typing:
Clustered at 98% nucleotide identity, leaving 161 representative sequences of all sero and genotypes.
DEN-IM
| A case study
Paired-end short-read Illumina shotgun sequencing dataset:
9 plasma samples
13 serum samples
1 spiked sample with the 4 serotypes
Positive and Negative controls
Paired-end short-read Illumina shotgun sequencing dataset:
Pipeline execution with default parameters and resources
DEN-IM
| The report
DEN-IM
| The report
DEN-IM
| Main points
https://github.com/B-UMMI/DEN-IM
https://github.com/B-UMMI/DEN-IM/wiki
This work was funded by: FCT - "Fundação para a Ciência e a Tecnologia" (SFRH/BD/129483/2017) and the Abel Tasman Talent Program grant from the UMCG, University of Groningen, Groningen, The Netherland
https://doi.org/10.1101/628073
Special thanks to E Lizarazo, M P Machado, D N Silva, A Tami, M Ramirez, N Couto, J W A Rossen, J A Carriço
By Inês Mendes
Slide presentation for the Bioinformatics and Computational Biology Seminars (10/07/2019)



